回答:
首先你需要代表性批次的稳定性方案和数据可以支持36个月的有效期,并按照公司内部的变更控制体系发起变更。
FDA
A. 重大变更(需事先批准的补充申请)
下面是一些被认为有重大可能性对药品的鉴别、规格、质量、纯度或效价等可能与药品的安全性或有效性相关的因素具有不良影响的变更例子。
1. 需要按照21 CFR第320部分完成研究以证明变更后的药品与未变更前生产的药品或指参照是等效的变更(§314.70(b)(2)(ii))。
2. 增加稳定性方案或可比性方案。
3. 对已批准的稳定性方案或可比性方案的变更,本指南中另有规定的除外(例如,VIII.C, VIII.D, XI.C.2)。
4. 基于(1)未在申请中获批的新的或修订的稳定性试验方案而获取的数据,或(2)采用已批准的方案获得的关于中试批次的完整货架期数据,而延长有效期。
5. 由于有关支持申请的数据可靠性的重要问题,对需要进行有效性评估的申请中的产品的变更(§ 314.70(b)(2)(viii))。
EMA
对于不同程序获批的产品,申请人申报或咨询的对象不同:对集权程序(CP)获批产品,应向EMA递交申请;对仅在某成员国(NP)获批的产品,应向该成员国药监局递交申请;其他类别(DCP/MRP)获批的产品,则向参考成员国(Reference Member State,RMS)和相关成员国(Concerned Member State , CMS)递交申请。
- Type IA 微小变更:对药品的质量、安全性和有效性影响很小或没有影响。 此类变更应在变更实施后的12个月内,向所有有关监管部门提交通知(annual notification (AN));但对于持续监管的需要立即通知的药品,需要在变更实施后立即提交通知(immediate notification (IN))。
- Type II 重大变更:不是Extensions,可能会对药品的质量安全性和有效性有重大影响。对于DCP/MRP获批产品,持有人同时向所有有关监管部门提交通知,RMS监管局应确认收到有效申请并知会持有人以及其他相关监管局:该程序从知会之日起开始;60天内,RMS监管局应准备好评估报告和对于该申请的决定,并传达给相关监管局;RMS可根据事件的紧急性缩短时限,延长适应症变更/新增适应症或组合变更的时限。 对CP/NP获批产品,则分别为EMA和相应成员国药监局。(major (MAJ))
- Type IB 微小变更:既不是Type IA型,也不是Type II型重大变更,也不是Extensions的。 对于DCP/MRP获批产品,应向有关监管局提交通知,RMS监管局与其他成员国协商后,确认收到了有效的通知,之后30天内,持有人未收到不利意见,则视为接受该变更通知;持有人收到不利意见的30天内,可提交修订通知,RMS在收到通知后30天内评估。对CP/NP获批产品,则分别为EMA和相应成员国药监局。(minor (MIN))
- Extensions:某些原料药的变更;规格、剂型和给药方式的变更;食用型动物用药的某些变更,或添加目标物种。该类上市许可应按照初始上市许可程序进行。
- CEP
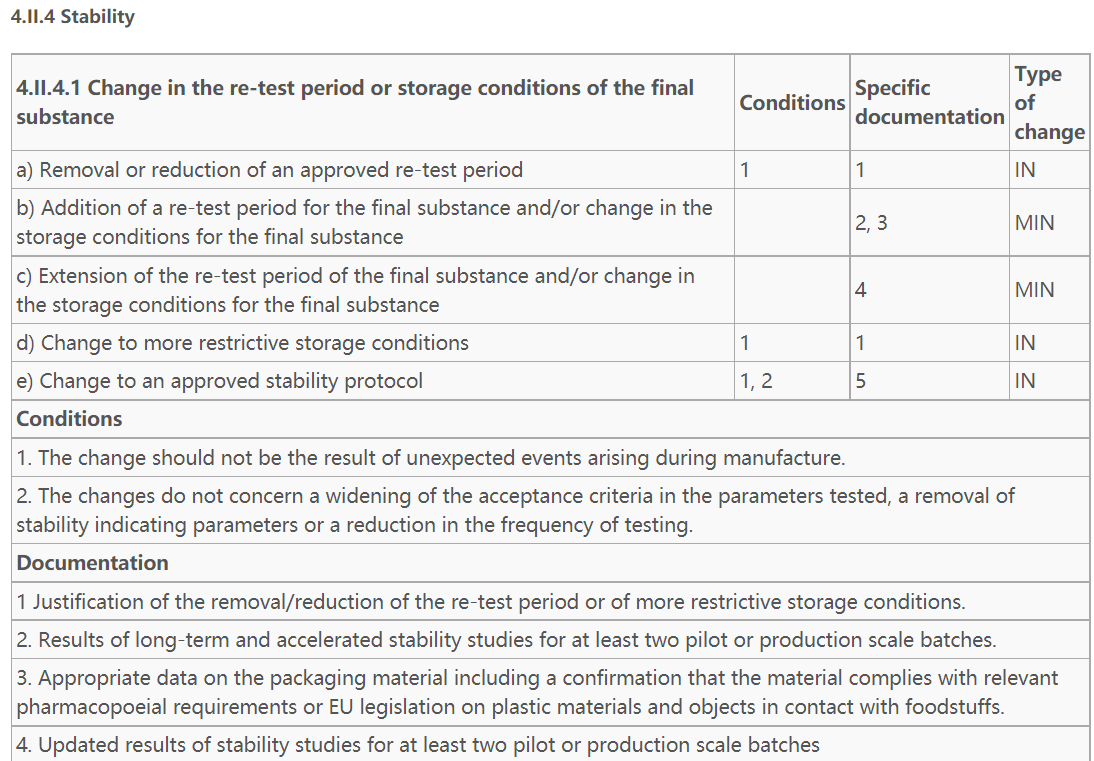
- 参考:
- CEP的要求
- 已获批NDA、ANDA变更行业指南
1、对于CEP
1)EDQM发布的PUBLIC DOCUMENTPA/PH/CEP (04) 2,对变更类型、支持资料等进行了详细说明,可参阅。根据该指南4.II.4.1 Change in the re-test period or storage conditions of the final substance项下c)Extension of the re-test period of the final substance and/or change in the storage conditions for the final substance,为微小变更,提供至少两个中试或生产规模样品更新后的稳定性数据即可。
需要注意的是,如延长效期是通过数据外推,要关注数据变化趋势,如有指标有增加趋势,会被挑战;此外,此类情况下即使提交36个月稳定性数据,但对应指标0月时实测结果较低,也是不能被接受的,因为无法保障任意批次产品延期后效期内一定合格。
2)根据EDQM发布的PUBLIC DOCUMENT PA/PH/CEP (13) 110,第一轮审评时限约1个月。
3)CEP证书版本号会更新。
4)提交前建议通知客户,拿到新版本号的CEP后及时签发新的授权信给客户。
2、对于US DMF
1)数据趋势问题同上。
2)提交前应通知客户,提交后应签发新的授权信给客户。
3)提交一个Amendment即可(可以在Cover letter里说明已通知客户及通知客户的时间)。通常更新资料为稳定性数据和标签样稿。
4)通常FDA不会有任何反馈。如有新的客户引用该DMF或原客户提交了补充申请,会在给客户的缺陷信中说明引用的DMF会进一步评估是否充分。如有问题会再次给DMF Holder发缺陷信,按照要求答复就好;如无问题,没有信息反馈给DMF方,不影响制剂的批准。
另:对于原料药,欧美一般不适用有效期,通常表述为复验期。
以上供参考,希望对你有帮助。
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: