根据CDE指南对于非无菌产品关注生产过程的微生物负载研究,对于能够降低微生物负载的工艺,可用下降对数值证明工艺除菌能力,而对于能能提高微生物负载,一般情况下,不会设置对比标准,而是通过设定内控标准(比法定标准更严格来控制,通常微生物检测值不符合正太分布,因此采用非参数方法设定内控警戒标准),在工艺时限内都必须满足该内控标准。
举个例子:比如中间产品存在时限验证,中间产品微生物限度标准(<10cfu),那在0时和72时检测样品均得满足该标准。
国家药监局药审中心关于发布《非无菌化学药品及原辅料微生物限度研究技术指导原则(试行)》的通告(2023年第11号)
4、制剂生产过程中的微生物控制 可从水分活度、生产工艺、中间产品存放时限和其他因素等 多个方面关注制剂生产过程中的微生物控制。
4.1 水分活度 非固体剂型(例如溶液剂、混悬剂、洗剂、乳膏剂、软膏剂 和凝胶剂等)比固体剂型具有更高的水分活度,支持微生物生 长的风险更高。水分活度是非无菌药品的重要属性,需关注原 辅料自身水分活度、吸湿特性、环境条件(温度、湿度)、贮存条件、包装系统等对水分活度的影响。 对于固体制剂、非水性基质液体制剂,水分活度较低时,微 生物不易生长和繁殖,但制剂初始生物负载仍可能在有效期内 持续存在,因此,对原辅料进行合理的微生物控制较为重要。
4.2 生产工艺 某些工艺步骤可能在提高或降低生物负载方面有较大影响。 应结合生产工艺的评估情况,对有微生物污染或生长繁殖风险 的关键工艺步骤,如水分活度较高的工艺步骤(配液、制备包衣 液等)或工艺时限较长的工艺步骤进行生物负载的研究。也可对能够降低生物负载的工艺步骤进行研究,通过研究证明微生物下降的程度(通常用下降对数值来表示),将有助于对工艺的 理解和工艺参数的控制。
4.3 中间产品存放时限 应对中间产品(如,可能更有利于微生物生长和繁殖的水性 基质中间产品)的包装形式、存放时限及环境条件进行充分研 究。必要时,应制定中间产品生物负载的限度标准,以监控工艺 中微生物数量和种类变化,保证其生物负载可控。
4.4 其他因素
生产过程中应减少由环境、设备、水系统、气体、清洁和消
毒剂、人员等因素引入的微生物污染风险。
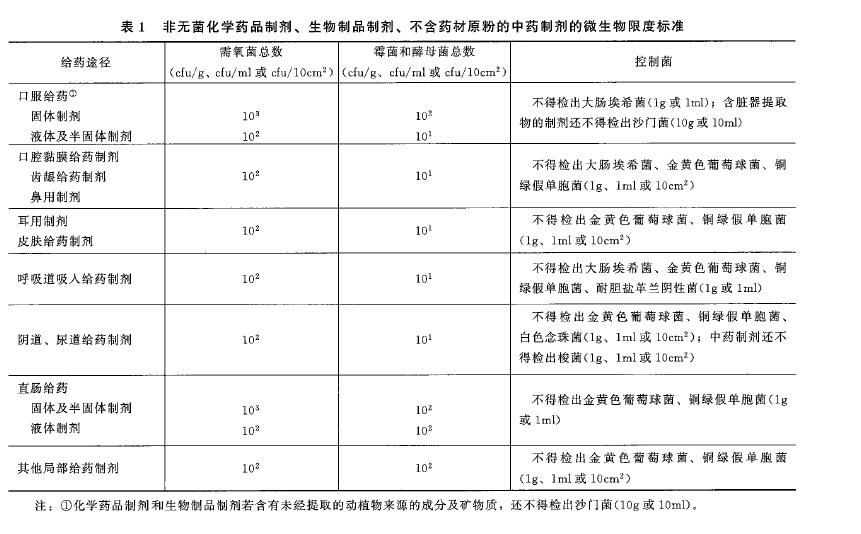
1. 对于口服液体制剂的微生物限度标准,参考中国药典1107 非无菌药品微生物限度标准:需氧菌总数100,霉菌和酵母菌总数10,不得检出大肠埃希菌。此外,值得注意的是,2023年2月CDE发布的指导原则(同下文)提出对洋葱伯克霍尔德菌群的控制,“对于吸入用途的非无菌制剂以及口服、黏膜、皮肤和鼻腔给药的水性基质非无菌制 剂,应参照相关技术要求对洋葱伯克霍尔德菌群进行风险管理和控制研究,制定相应的控制策略,必要时将洋葱伯克霍尔德菌群订入产品放行/注册标准(登记标准)”,应开展相关的研究。
2. 对于制剂灌装过程中的微生物限度控制,可参考CDE发布的《非无菌化学药品及原辅料微生物限度研究技术指导原则(试行)》的通告(2023年第11号)https://www.cde.org.cn/main/news/viewInfoCommon/b522b0ea49412b5edc52f002a1d1036a
“中间产品存放时限
应对中间产品(如,可能更有利于微生物生长和繁殖的水性
基质中间产品)的包装形式、存放时限及环境条件进行充分研究。必要时,应制定中间产品生物负载的限度标准,以监控工艺中微生物数量和种类变化,保证其生物负载可控。”即可以进行充分的存放时限研究后,根据研究结果制定合理的过程控制标准,以确保最终制剂产品的微生物限制标准可控。
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: