依据《中华人民共和国药品管理法》与药物警戒相关的内容:
总则 第十二条明确要求建立药物警戒制度,对药品不良反应及其他与用药相关的有害反应进行监测、识别、评估和控制。
第三章 药品上市许可持有人 第三十条要求,持有人对药品的上市后研究、不良反应监测及报告与处理等承担责任。
第七章 药品上市后管理 第七十七条规定,持有人应当制定药品上市后风险管理计划,主动开展药品上市后研究,对药品的安全性进行进一步确证;
第八十条规定,持有人应当开展药品上市后不良反应监测,主动收集、跟踪分析疑似药品不良反应信息,对已识别风险的药品及时采取风险控制措施。
第八十一条规定,持有人应当经常考察本单位所生产药品的不良反应,发现疑似不良反应的,应当及时向药品监督管理部门和卫生健康主管部门报告。
第八十三条 药品上市许可持有人应当对已上市药品的安全性、有效性和质量可控性定期开展上市后评价。必要时,国务院药品监督管理部门可以责令药品上市许可持有人开展上市后评价或者直接组织开展上市后评价。经评价,对疗效不确切、不良反应大或者因其他原因危害人体健康的药品,应当注销药品注册证书。
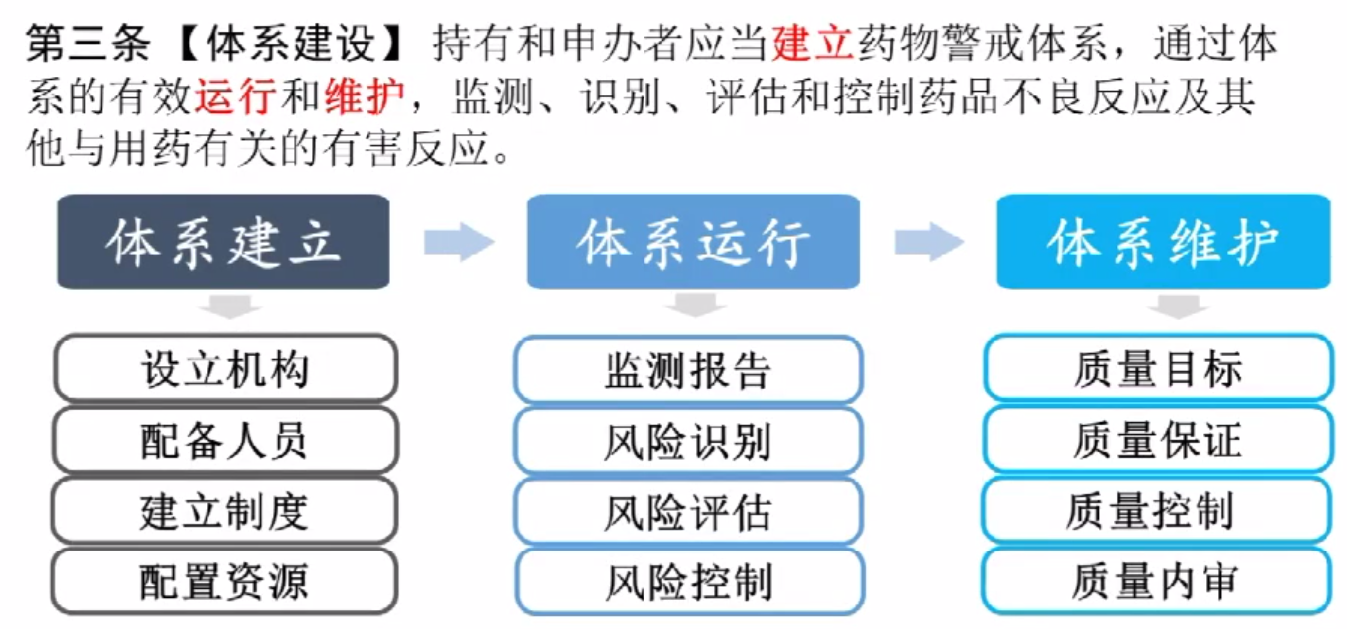
需要根据药物警戒管理规范,建立药物警戒体系,依据法规要求和企业的实际情况完善公司药物警戒体系。总体框架如下:
总则-管理规范的制定依据、适用范围、总体原则等
药物警戒体系与质量管理-持有人应当建立药物警戒体系,创建并维护体系文件,对质量管理提出总体要求
机构和人员-对药物警戒体系的组织机构、人员和开展药物警戒工作所必须的资源提出具体要求
安全性信息监测与报告-规范个例药品不良反应报告、定期安全性更新报告和药品不良反应聚集性事件的处置和报告;
风险识别与评估-持有人如何发现、评估药品风险;
风险控制和沟通-风险控制措施的总体要求和集体形式
上市后安全性研究-规定上市后安全性研究的总体要求,对重点监测提出原则性要求;
文件和记录管理-规范药物警戒或相关文件和记录的管理;




这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: