【依据】以下对我国突破性治疗药物、附条件批准、优先审评审批等加快上市注册程序的对比介绍
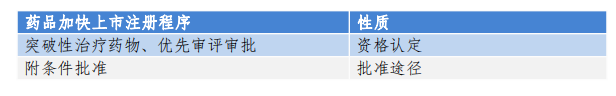
1 药品加快上市注册程序的性质
2 法规来源

3 适用范围

4 何时递交申请

5 CDE 回复的时间表

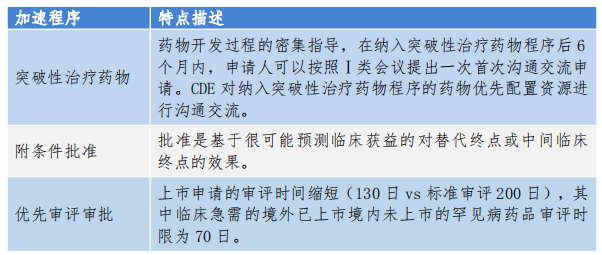
6 特点
7 其他注意事项

【思考】
首先,突破性治疗药物、附条件批准、优先审评审批等加速上市程序是相互独立的,例如申请附条件批准的药物,不一定要获得突破性治疗药物资格,需要根据药品开发情况、临床数据和注册策略,进行选择或组合。获得突破性治疗药物资格的药物,(1)如果能够获得附条件批准,则基于早期临床结果加速上市,同时叠加优先审评审批,缩短审评时限(200日变130日);(2)如果不满足附条件批准,也可申请普通的上市审批,叠加优先审评审批。
其次,这三种程序,只是加速了药物的上市,但绝不是允许降低药品研发质量水平,也绝不是可以豁免临床研究。以附条件批准为例,(1)申请附条件批准上市的药品的药学、药理毒理学要求与常规批准上市药品相同,这一点不能有任何的含糊。(2)临床试验设计和执行过程不能存在缺陷。特别注意应先有严谨、科学的临床试验,并获得部分结果,经讨论、评判后才有可能“附条件批准”,而不是为了“附条件批准”去设计相应的临床方案。(3)临床疗效指标的选择科学合理,能够保证其很有可能预测临床获益,并早于临床终点进行测量,这是“附条件批准”最重要的指标,所以临床研究的主要终点、次要终点以及替代指标的设置,需要全面评估,以期多角度反映候选新药的疗效。(4)已获得的安全性和有效性数据,能够表现出明显优于现有治疗手段的临床价值,即获益大于风险。(5)附条件批准不是完全批准,药品获得附条件批准上市后,持有人应按照规定完成上市后研究。一是必须如期完成确证性临床试 验来评价药品临床获益。二是收集药品安全性数据,定期评估与药品使用相关的风险情况。当持有人按 期提供完整的、能够证明临床获益的临床数据时,附条件批准上市的药品可申请常规批准上市。若临床终点结果不能证明获益大于风险,或持有人未按期完成确证性临床试验时,监管机构将暂停或撤销附条件批准。
【案例】
江苏豪森的甲磺酸阿美替尼片,CDE 建议附条件批准该药品上市,而适应证的完全批准将取决于正在进行的Ⅲ期确证性随机对 照研究( HS-10296-03-01 ) 所证实的临床获益。
又如江苏恒瑞的马来酸吡咯替尼片,基于在HER2阳性晚期乳腺癌中开展的吡咯替尼联合卡培他滨的I/II 期临床研究(HR-BLTN-I/II-MBC)结果,申报生产,并获得附条件批准。在其上市后要求中,CDE提出:
(1)请按计划进行完成正在进行的2项Ⅲ期研究(HR-BLTN-Ⅲ-MBC-A研究和HR-BLTN-Ⅲ-MBC研究)。按年度报告其进展情况,完成的研究应及时提交完整的总结报告。
(2)针对HER2表达阳性,既往未接受过曲妥珠单抗的晚期或转移性乳腺癌患者,目前获得数据有限,应开展一项随机对照研究确证本品在该人群的疗效。在批准上市后3个月内提交完整方案和实施进度时间表。
(3)药代数据显示吡咯替尼存在较大的个体差异,在体内广泛代谢,疾病背景、肝肾功能、年龄、合并用药等有可能影响药物暴露水平。建议后续在更大样本中收集PK数据,建立适宜模型,评估可能影响暴露的因素,如性别、年龄、肝功能不全等,以更好指导临床用药。
(4)请适时开展药物相互作用研究,包括肝药酶诱导剂和抑制剂对本品药代影响的研究、胃酸分泌药物对本品药代影响的研究、本品对P-糖蛋白底物(如地高辛)代谢的影响研究。
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: