这个问题,我之前也好奇过,搜过,规定清楚一个责任主体就行,总得有个负责人,不能让监管找不到人;至于实际干活,可以委托给外面公司去做,我公司现在总部,部分项目跟多个CDMO合作,部分项目由子公司跟CDMO合作,总公司和子公司之间也有部分关联。
此话题,再进一步延伸,那就是临床期间是否允许多少个申办者(当然性质不一样,只是思维发散),上市后又允许多少个持有人?主要起因是CDE在2020年04月15日发文“药审中心关于药品上市注册申请确认持有人有关事宜的通知”,由此衍生的,既然国内的,上市后规定只能有一个,那国内临床期间的呢,若继续延伸到欧洲和美国呢?思维进行发散学习。具体详情见下:(国内在上市前和上市后1.1+1.2的截图;欧洲在上市前和上市后2.1+2.2的截图;美国在上市前和上市后3.1+3.2的截图);
【先说结论:①只有欧洲允许在上市前有多个,但上市后只允许1个,哪怕是集团内的也得视为同一个;②FDA明确上市前和上市后只允许1个;③国内在上市后只允许1个,上市前暂无法规支持与明确,得看官方解释和线下实操情况,感兴趣的,详情见下截图原文】
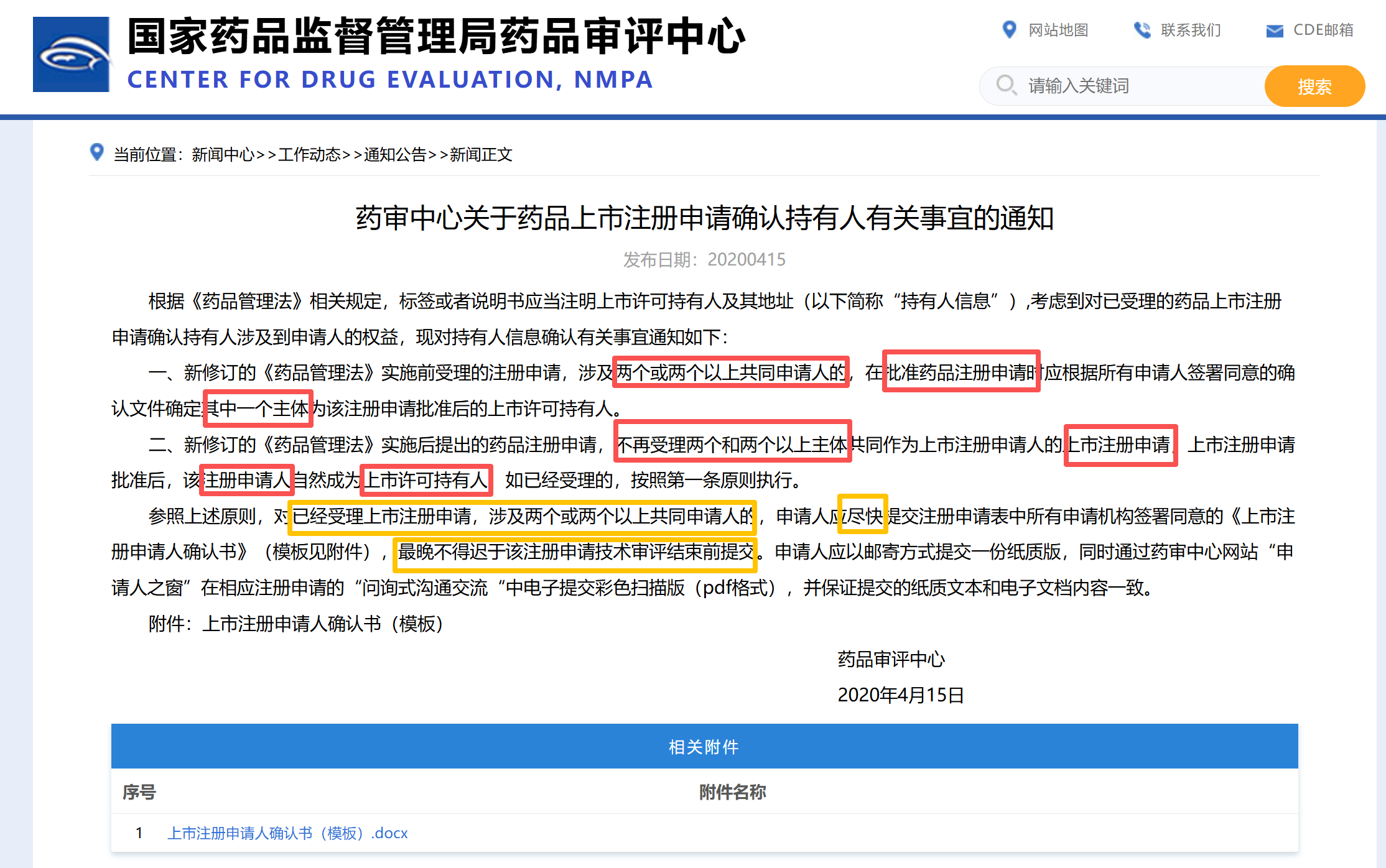
1.1:国内在上市后明确只能1人,如下图:
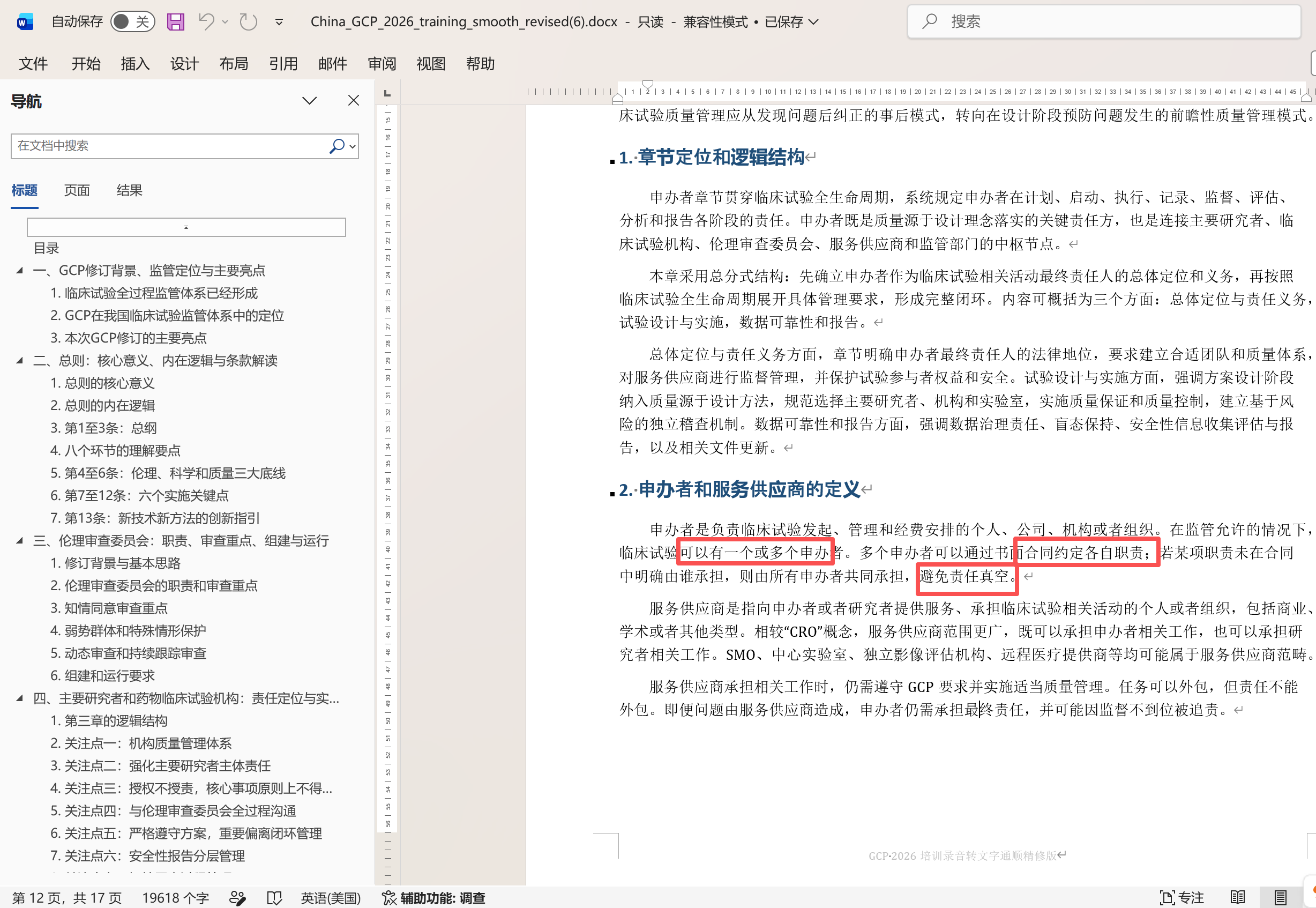
1.2:国内在上市前的,这次07.02的官方培训新版的GCP,明确了,允许是多个,特此来修订答案,详见以下官方课件和截图:

但是欧洲在上市前允许多个,在上市后只允许1个(美国在上市前只允许1个,上市后也只允许1个)的截图如下(网上公众号看到的法规截图原文,我核实了法规的现行版,这个倒没错,但因为我不是专门研究临床的,若必要时,对临床期间发文再细查,之所以贴出来,因为作为QA一般只相信法规原文,非法规原文的只能半信半疑,可能是部分实操的特殊情况,那只能靠经验了)
2.1:欧盟在上市前(允许多个):《REGULATION (EU) No 536/2014 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 April 2014 --on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC》欧洲议会及理事会 2014 年 4 月 16 日第 (EU) 536/2014 号法规—— 关于人用药品临床试验,并废止第 2001/20/EC 号指令


2.2:欧盟在上市后(只能是1个,哪怕是集团公司的,也只能视为1个):有EMA官方的问答(Questions and Answers on List for the submission of variations for human medicinal products According to Commission Regulation (EC) 1234/2008 as amended根据修订版欧盟委员会法规 (EC) 1234/2008 提交人用药变更的问答清单),还有之前做课件的定义(去找的法规原话),如下两张截图:


3.1:FDA在上市前(只允许1个申办者sponsor):
IND申请表(Form FDA 1571)

3.2:FDA在上市后(只允许1个申请人/持有人),是因为美国需要指定“唯一代理人”:


这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: