对于注射剂上市后增加规格的核心审评逻辑是“立题依据的合理性”,即使是化药新药增加规格也应当考虑。
(1)是否符合临床需求:是否有明确的适用人群(如儿童、特殊病种)、是否能解决现有规格的使用痛点(如配液麻烦、浪费严重、剂量误差大);
(2)是否在合规范围内:是否在说明书用法用量范围内,是否小于单次最小用量或大于单次最大用量;
(3)是否有数据支撑:是否提供临床必需性的文献/数据支持,而非企业主观意愿。
若该化药新药小规格的上市是为了满足特殊用药人群的需求,例如儿童,需提供儿童人群的安全有效性数据,或桥接原规格的临床数据;
建议:立题阶段先沟通交流,通过沟通交流向CDE明确小规格的临床价值、用法用量合理性,避免立题不通过。
以下是案例分享:
一、体积不变+降低浓度--成功案例,但时间较久,不一定符合最新审评尺度。
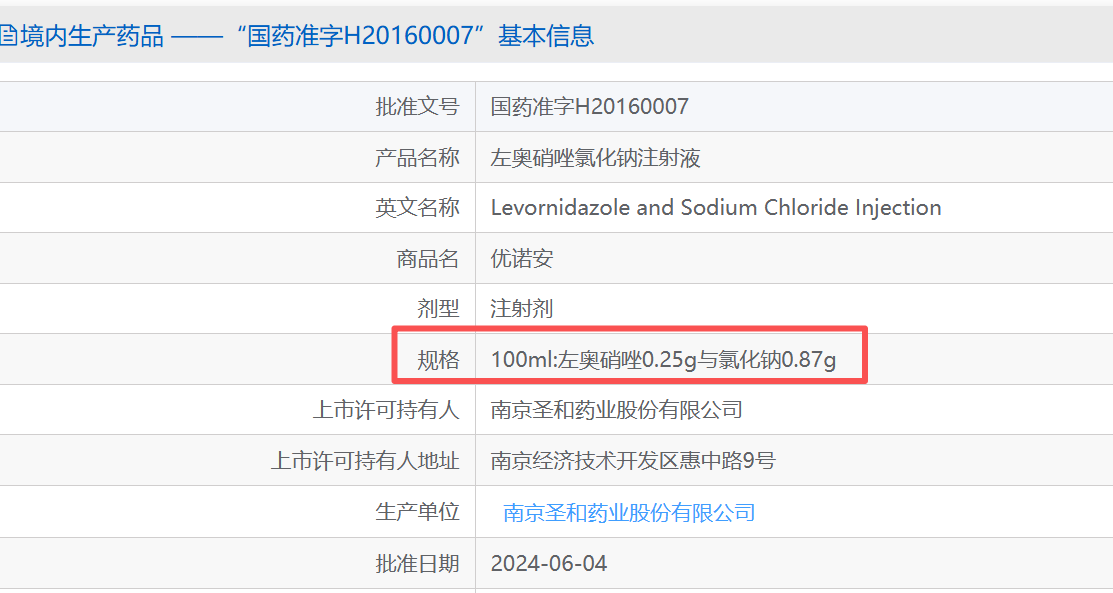
南京圣和药业原研左奥硝唑氯化钠注射液2009年按1.3类获批规格为100ml:左奥硝唑0.5g/氯化钠0.83g,2016年成功补充申请增加100ml:左奥硝唑0.25g/氯化钠0.87g。

二、仿制药由于规格合理性问题不批准案例
1、胞磷胆碱钠注射液:公布的参比制剂规格为4ml:500mg和4ml:1g;山东齐鲁制药有限公司申请了原有品种规格2ml:0.25g的一致性评价未获批。
2、注射用更昔洛韦:公布的参比制剂规格为0.5g,武汉人福申报的0.25g和0.5g两个规格均通过一致性评价,而申报的0.05g的规格一致性评价未通过。
3、注射用阿奇霉素:参比制剂的规格为0.5g,而国内原有规格中有0.125g(25个文号),0.25g(50个文号),在进行一致性评价时,由于两种规格小于
临床单次使用的最小剂量,而不被批准。
药监局在2004年发过一个通知“关于加强药品规格和包装规格管理的通知”,对于规格部分的要求主要如下:
一、由于药品的规格及用法用量是依据上市前临床试验确定的,因此,申请增加的规格一般应当与同品种上市规格一致。
二、申请增加的药品规格与同品种上市规格不一致的,必须符合科学性、合理性、必要性原则,必须经过充分的论证,以免给临床合理用药造成混乱。
三、申请增加的药品规格不得改变原用法用量或者适用人群。
四、申请增加的药品规格应当根,据药品用法用量合理确定,一般不得小于单次最小用量,也不得大于单次最大用量。
五、除按照上述规定执行外,大输液一般不受理50ml、lOOml、250ml、500ml以外的其他规格,水针一般不受理1m1、2ml、5ml、l0ml、20ml 以外的其他规格。
1.基本原则:药品制剂规格需结合药代动力学、药效动力学、人体机体情况及用药便利性综合科学制定。
2.硬性要求:新增/变更规格必须适配原有批准的用法用量,同时兼顾临床与患者用药便捷性;不能出现规格偏大、偏小、剂量不匹配、浓度改动后无法按原用法使用等问题。
3.立题依据不足的情形:包含规格大于/远小于单次用量、剂量/浓度改动后无法按原用法使用、改规格却擅自变动适应症和用法用量且无临床研究支持这五类,难以获批。
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: