答主引用曾子像老师 药理毒理开发公众号推文“支持药物IND和上市申请的非临床给药周期设计”https://mp.weixin.qq.com/s/6s46hw3__X_Sc_wQlKipTg
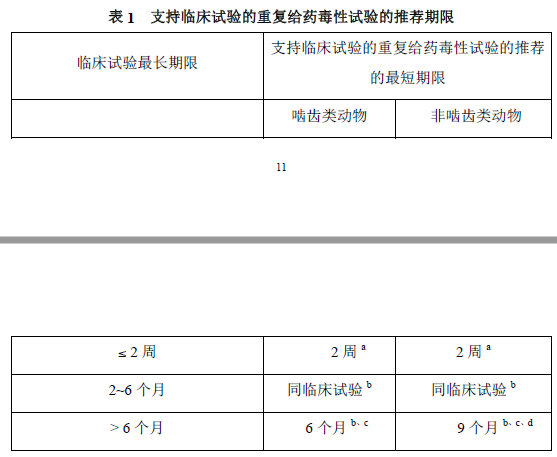
先看下ICH M3重复给药毒性研究部分对于给药周期的要求。首先是支持临床试验的要求。重复给药毒性试验给药周期有上限和下限。下限是,对于临床持续周期≤2周,重复给药毒性试验则至少要持续2周。上限是,对于临床持续周期>6个月,重复给药毒性试验倒不必一定与临床一致,啮齿类动物满足6个月,非啮齿类给够9个月即可。对于2周-6个月之间的情况,与临床周期一致即可。
也有些特殊情况特别说明一下。比如在美国,临床仅单次用药,非临床研究可以采用拓展的单次给药毒理研究设计。对于临床研究持续<14天的情况,非临床给药周期可以与临床保持一致。又如在欧洲,通常6个月非啮齿类毒理研究可以支持临床>6个月。在日本和美国,如果出现以下情况,6个月也可以作为最长非啮齿类给药周期,比如免疫原性或动物不耐受影响更长周期毒理试验的正常开展,或间歇性治疗偏头痛、勃起功能障碍或单纯疱疹这种短期重复暴露的情况,或慢性用药为降低肿瘤复发风险,或用于预期寿命比较短的适应症。
生物制品临床前安全性评价可参考ICH S6。给药途径和方案(如每天给药vs间隔给药)应该反映临床拟定使用或者暴露情况。关于重复给药毒性试验的期限,应根据预期临床暴露的期限和适应症确定。大多数生物制品的动物给药期限为1-3个月。对于计划短期使用(如≤7天)或者治疗急性危及生命疾病的生物制品,2周的重复给药试验可以支持其临床试验以及上市许可。对于拟用于慢性适应症的生物制品,试验期限一般为6个月。
抗肿瘤药物非临床评价参考ICH S9。抗肿瘤药物通常采用的非临床试验给药方案的实例,可应用于小分子药物或生物药物。下表展示了支持早期临床试验的非临床给药方案设计。按照这个表格,如果临床每3-4周给药1次,非临床开展单次给药毒理试验即可支持IND,但也不绝对。以ADC为例,临床多是Q3W给药频率,ICH S9问答中对于这类产品,建议至少给药2次。
另外,关于支持后续临床试验和上市的毒理学研究期限。支持Ⅰ期临床试验的非临床资料和Ⅰ期临床资料通常足以支持进行Ⅱ期临床试验,和支持进入晚期肿瘤患者的二线或一线治疗。为了支持用于晚期肿瘤患者抗肿瘤药物的后续开发,在Ⅲ期试验开始之前应提供符合临床拟定方案的为期3个月的重复给药毒性试验结果。对于拟用于晚期肿瘤患者的大多数药物来说,为期3个月的非临床毒性试验足以支持上市。
生物药和化药的毒理试验有一定的区别,同时根据适应症的不同,毒理试验的周期要求也有一定的不同。一般其它试验周期都不超过长毒试验的周期。
在IND(新药临床试验申请)阶段,毒理实验主要包括:
1、急毒试验 (通常需啮齿类+非啮齿类动物)
2、DRF试验(剂量探索性重复给药毒性试验,也可不进行,但会增加长毒试验风险)
2、 重复给药毒性 (长毒试验,化药一般需2个相关种属,啮齿类+非啮齿类动物,生物制品可选一种相关种属,一般为猴),给药周期要求需参考临床试验周期要求,可参考ICHM3(R2)
3、安全药理学
4、遗传毒性 (化药:Ames+染色体畸变+体内微核试验,生物制品一般不需要)
5、生殖毒性 (一般IND阶段不会进行)。
在IND(新药临床试验申请)阶段,毒理实验需根据药物类型(化药/生物制品)、适应症及监管要求设计,核心内容包括 急性毒性 (单次给药评估最大耐受剂量,通常需啮齿类+非啮齿类动物,豁免情况除外)、 重复给药毒性 (覆盖临床疗程,化药需两种动物,生物制品可选一种相关种属)、 安全药理学 (评估心血管、中枢神经及呼吸系统影响)、 遗传毒性 (Ames试验+哺乳动物细胞试验+体内微核试验,生物制品可能豁免),以及 生殖毒性 (早期临床可暂缓完整试验,肿瘤药按ICH S9延迟)。此外,生物制品需关注 免疫原性 和靶点毒性,基因毒杂质需按ICH M7评估并控制。试验需符合GLP规范,时间因项目而异:化药通常需6-12个月(遗传毒性3-6月,重复毒性3-6月),生物制品若种属明确可缩短至3-9个月,具体受模型建立、剂量探索及监管沟通影响。
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: