5个回答
建议密封性测试需放在放行标准中,每批检测放行,无菌检测仅是微生物方法(概率)。
参考《关于无菌药品包装系统密封性指导原则标准草案的公示(第二次)》2024-06-21关于密封性检测方法的说明,在产品生产阶段,推荐要求在线和离线均需进行密封性检测。
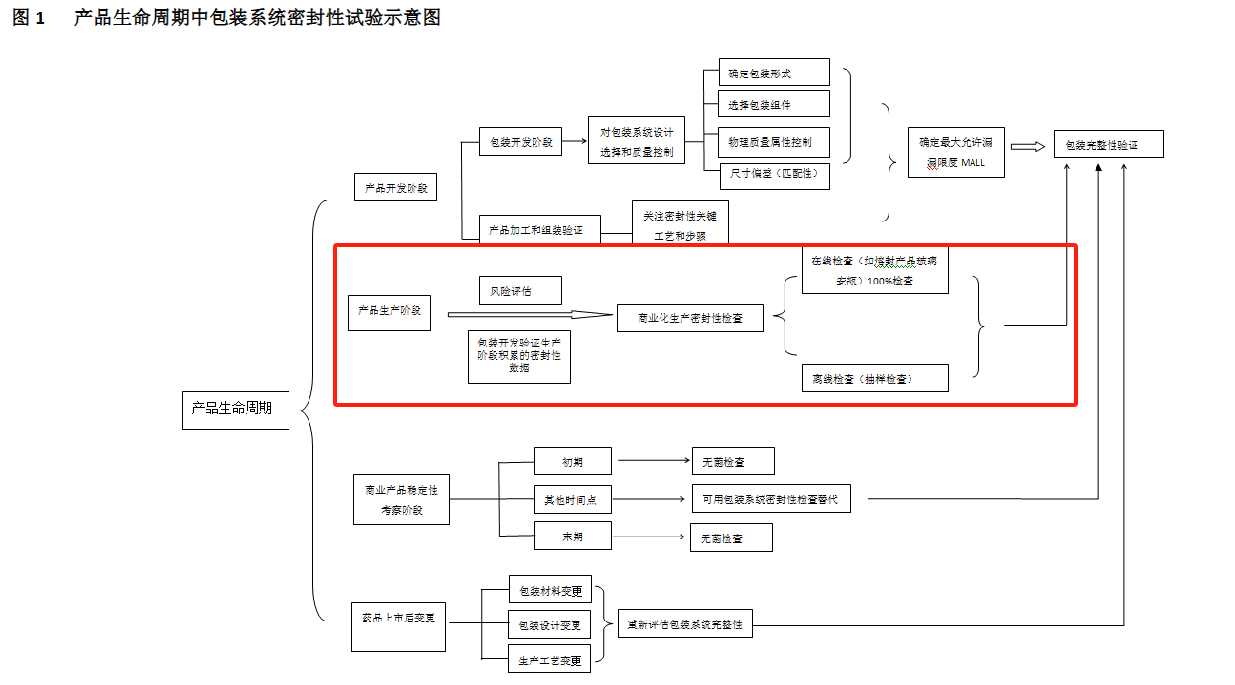
三、产品生命周期中包装系统的密封性研究对包装系统密封性的考察和监测应从产品风险管理角度出发,贯穿产品的生命周期(见图 1)。包装系统密封性的保证不是仅仅通过最终产品抽样检查或在线检查控制,还与包装系统/组件的设计、选择、生产,产品生产过程控制,贮藏运输管理等有关。从包装系统用途出发,在包装系统/组件的开发和加工及组装验证阶段,需确定包装系统的固有包装完整性,基于质量源于设计(QbD)的理念,应关注组件选择的匹配性以及工艺参数的设定和优化;在产品的生产阶段,当产品、包装设计、包装材料或生产/加工条件发生变更时,需考虑密封性的重新评价并在变更实施前完成风险评估;在药品稳定性考察初期和末期进行无菌检查,其他时间点可采用包装系统密封性检测替代,监测产品贮存过程中的质量变化。在产品生命周期中的各个阶段不断积累密封性评估和控制的知识、历史数据和经验,制定有效的密封性控制策略,有利于持续可靠地确保密封性符合预期要求。
评论
匿名
提交
取消
匿名
{{item_parent.created_at}}
置顶
批准
驳回
编辑
等待审核
已驳回
回复
{{item_parent.show_reply_list ? '收起回复' : '查看回复'}}({{item_parent.children.length}})
编辑
提交
取消
写回复
匿名
提交
取消
{{item_children.from_user}} 回复 {{item_children.to_user}}
{{item_children.created_at}}
批准
驳回
编辑
等待审核
已驳回
回复
编辑
提交
取消
写回复
匿名
提交
取消
这{{threadTextType}}正{{isAdminText}}
举报
提交
取消
为帮助审核人员更快处理,请填写举报原因:
举报
提交
取消
为帮助审核人员更快处理,请填写举报原因: