作者|杜婧,董江萍(CFDI)
来源|中国医药导刊2021年第23卷第10期( 总第216期)
[摘要] 目的: 基于化学仿制药注射剂注册生产现场检查中存在的合规性问题和质量风险问题分析,提出质量管理建议,以期为药品监管部门监管工作及药品生产企业质量管理体系建设和完善提供参考。方法: 收集、整理我中心2019年度对68个( 以受理号计) 化学仿制药注射剂的注册生产现场检查工作实施情况,分析发现的主要问题。结果: 药品生产企业在无菌操作与人员培训、培养基模拟灌装试验内容、工艺验证和确认、共线生产风险评估及清洁验证等方面的GMP符合性问题发生率高。结论: 对于药品监管部门,建议对注射剂生产企业的薄弱环节进行重点监管; 对于注射剂药品生产企业,建议应严格按照相关法规要求,进一步提高无菌保证能力,全面提高我国仿制药注射剂的质量,充分保证药品的安全、有效和质量可控。
注射剂( injection) 系指药物制成的供注入体内的无菌溶液( 包括乳浊液和混悬液) 以及供临用前配成溶液或混悬液的无菌粉末或浓溶液,是临床上一种常用制剂,在临床治疗中占有极其重要的地位。
我国医药市场中,化学药品注射剂是注射给药剂型的主要品种,其市场规模占全国注射剂整体市场的72%左右。有研究报道,化学药品注射剂市场近5年
呈现不断增长趋势,已由2013年的4085亿元增长到2018年的6152亿元。化学仿制药注射剂在药品市场中有着举足轻重的地位。
从使用角度,注射剂因属于直接注入人体的给药剂型,相对于口服制剂风险更大。据原国家食品药品监督管理总局 (CFDA) 发布的2016年度
《国家药品不良反应监测年度报告》显示,2016年
药品不良反应/事件报告中静脉注射给药占59.7% ,其他注射给药( 如肌内注射、皮下注射等)
占3.4%。2020年《国家药品不良反应监测年度报告》显示,静脉注射给药占91.1% ,其他注射给药占8.9%。注射剂本身的给药途径和临床应用范围决定了注射剂的高风险特点,通常被纳入高风险等级的产品强化监管。
注射剂在生产环节包含多个风险因素,生产过程中需采用无菌生产工艺,严格控制可能引起微粒、微生物和内毒素的潜在污染,对无菌生产工艺进行过程控制,确保无菌产品不受污染。所以对于注射剂产品来说,质量保证的重点在于无菌保证、细菌内毒素和微粒污染的控制,还需要特别关注防止混淆和交叉污染。
为加强新批准化学药品注射剂的质量安全,2018年5月国家药品监督管理局发布了《国家药品监督管理局关于加强化学仿制药注射剂注册申请现场检查工作的公告》( 2018年第 20号) ( 以下简称“20号公告”),专门组织对申请批准上市的化学仿制药注射剂开展生产现场检查。本研究通过对我中心2019年度根据20号公告启动的68个( 以受理号计)
化学仿制药注射剂注册生产现场检查工作的实施情况及发现主要存在的合规性和质量风险问题进行收
集、整理及分析,提出质量管理建议,以期为药品监管部门监管工作和上市许可持有人及药品生产企业质量管理体系建设和完善提供参考。
1 化学仿制药注射剂注册核查任务的基本情况
2019年度,国家药品监督管理局食品药品审核查验中心对68个受理号化学仿制药注射剂的品种开展注册生产现场检查。剂型分布方面,包括小容量注射剂、大容量注射剂及冻干粉针剂,按受理号计分别为24、6 和38个,占比依次为35% 、9% 和56% ( 见图1) ; 检查情形方面,首次申报的品种较少,按受理号计仅为2个,占比3% ,属于Ⅲ类变更 /
重大变更情形7个,占比10% ,生产地点( 生产线)
变更情形7个,占比10% ,绝大部分品种为Ⅲ类变更 /重大变更和生产地点(生产线) 变更同时开展,
品种数达到52个,占比77% ( 见图2) ; 各省申报情况方面,数量较多的申报省份包括山东、江苏、四川等。见图 3。
2 化学仿制药注射剂的特殊检查要求
鉴于注射剂的质量风险和生产工艺特点,年度展开的化学仿制药注射剂注射生产现场的检查重点包括注册申请人整体实施药品生产质量管理规范
(GMP) 水平与申报品种无菌保证能力,以及品种申报时动态生产批次情况,包括生产批量等与申报资料的一致性、真实性等相关内容。考虑到20号公告中涉及的检查范围均是注射剂,属于高风险品种,所以
此类型注册检查要点中除真实性、一致性、GMP符合性之外,还特别强调了申报品种无菌保证能力的要求。既往药品注册核查中,这是首次重点强调和要求
的内容。在完成的检查中,检查结论为不通过的有2个,占比3.5% ; 其余结论均为通过。
3 化学仿制药注射剂注册生产现场检查发现的主要问题
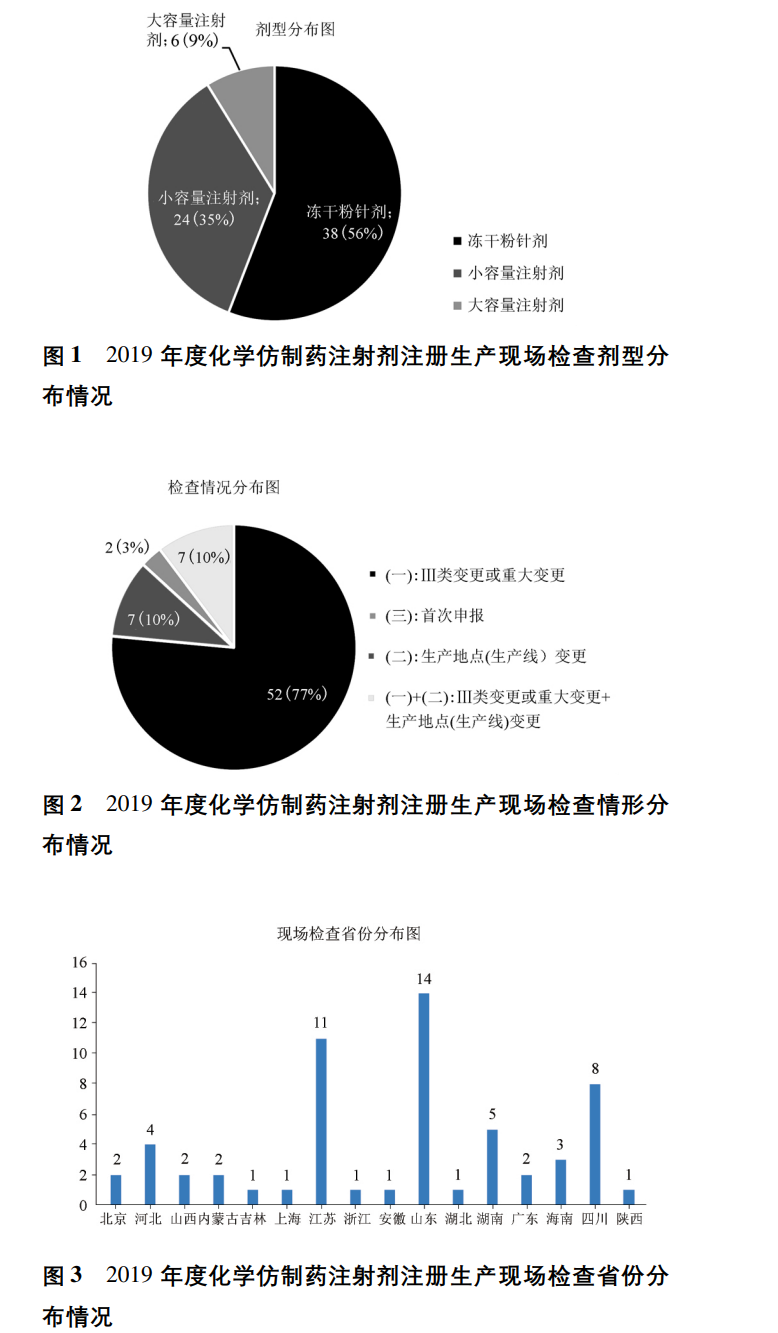
本研究发现2019年度化学仿制药注射剂注册生产现场检查中药品GMP缺陷共有682项,其中涉及药品GMP正文部分的缺陷524项,涉及计算机化系统附录的缺陷17项,涉及取样附录的缺陷6项,涉及确认与验证附录的缺陷28项,涉及无菌药品附录的缺陷107项。
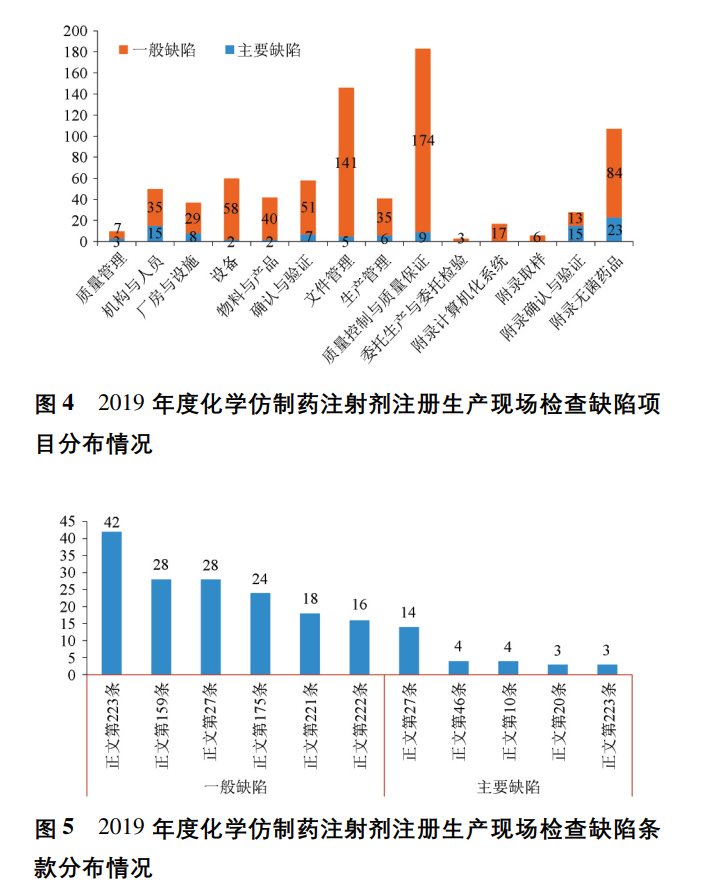
如图4所示,2019年度化学仿制药注射剂注册生产现场检查中质量控制与质量保证部分发现的缺陷最多,共183条,占全部缺陷的26.8% ; 其次是文件管理,共146条,占比 21.4% ; 再次是设备,占比8.8% 。本研究通过对提出的缺陷条款进行分析汇总,特别关注了提出频次较高的缺陷(见图5) ,研究发现主要缺陷中提出最多的条款是正文第27条( 人员培训) ,其次是正文第46条( 厂房与设施) 。一般缺陷中提出最多的条款是正文第223条( 药品检验) ,其次是正文第159条( 文件记录) 。
4 化学仿制药注射剂检查问题梳理和分析
经对注册生产现场检查所发现缺陷进行梳理,
2019年度注册生产现场检查未发现数据无法溯源以及申报资料不真实的问题。发现的问题主要聚集在GMP符合性方面,如无菌操作与人员培训不到位、培养基模拟灌装试验内容不完整、工艺验证和确认内容不充分、共线生产风险评估内容不全面及清洁验证不科学等问题。
4.1 无菌操作人员培训不到位
部分企业无菌操作人员培训不到位,无菌操作从事人员动作不规范,如灌装操作人员上身进入层流车区域进行无菌组装操作; 灌装、轧盖操作人员频繁进出A级层流区域操作等。上述操作容易对无菌区域产生污染,影响产品质量。
4.2 无菌工艺模拟试验内容不完整
部分企业无菌工艺模拟试验中,批生产记录内容不全,如轧盖机轧盖密封性检查各瓶号的样品无取样位置的记录; 无菌工艺模拟试验验证未通过风险评估来确定验证条件; 无菌检查的方法学验证报告中未记录验证结果和结论,记录内容不全面,追溯性不强。
4.3 工艺验证和确认内容不充分
个别品种工艺验证与确认工作部分内容不充分,
如灭菌工艺验证报告确认的产品装载方式与动态批灭菌装载方式不一致; 工艺验证时尚未完成除菌过滤器的验证,工艺验证方案及报告未把除菌过滤器的验证结果纳入考虑,也未进行相应的风险评估。
4.4 共线生产风险评估内容不全面,清洁验证不科学
部分企业对共线生产风险评估内容不全面,如新增品种和现有品种成分相似,认为不会产生污染和交叉污染,未将新增品种中的不同成分对现有品种可能产生的影响纳入共线风险评估,没有综合考虑药品的特性与预定用途等因素。个别企业共线生产细胞毒性药物和非细胞毒性药物,且厂房设施、设备、工器
具、风险控制措施、清洁验证不完善,存在污染与交叉污染的风险。清洁验证方面,清洁溶剂选择与产品的溶解性不相适应; 未对残留检测分析方法进行验证,
不能证明该方法的可靠性; 取样点、检测项目及标准规定不科学,也未基于毒理试验数据或毒理学文献资料确定限度标准; 未综合考虑阶段性生产的最长时间和最大批次数量,以作为清洁验证的评价依据。
4.5 部分环境监测项目、频次考虑不全面
部分企业洁净区域环境监测频次未能覆盖各品种生产过程,未规定悬浮粒子的动态检测周期。日常环境监测中,沉降菌监测点考虑不周全,未考虑环境监测数据的分析结果。检测记录中对采样点培养出的菌落数仅体现最终结果,未记录每天的观察情况,
所用设备型号、编号、设定参数等未体现,检测记录中的内容不全面。
5 建议
基于注册生产现场检查发现GMP符合性方面的问题,建议药品上市许可持有人履行生产质量保证的主体责任,药品上市许可持有人和药品生产企业应高度重视并重点加强以下几方面质量风险控制工作。
5.1 重点工艺环节生产人员的无菌保障意识
要加强无菌操作人员的培训,使操作人员熟知无菌工艺流程和各项操作规程,同时建立培训实操考核机制以及日常生产的监管机制,高风险区域可利用视频、录像等手段进行实时监控。加强操作人员的专业知识和无菌意识的培训。
5.2 无菌工艺模拟试验的科学性和合理性
建议根据产品的剂型特性选择合适的模拟介质开展无菌工艺模拟试验; 通过风险评估来确定灌装数量及模拟持续时间、容器装量、关键的模拟操作步骤及工艺参数,还应考虑生产中可能出现的固有干预、
纠正性干预和人员、时限、灌装速度等最差条件; 严格按照拟订的验证条件进行模拟灌装试验,并对试验过程中关键点进行详细记录。
5.3 工艺验证和确认工作的规范性
验证前应做好相关设备和方法的验证和确认,如无菌过滤器验证、无菌检测方法验证等准备工作; 生产工艺中关键工艺操作及其参数应在验证方案中明确,并确保在验证过程中准确无误的实施和记录; 对验证结果进行评估以评价工艺验证和确认的可行性。
5.4 多产品共线的风险评估与清洁验证方法的针对性
在共线生产前应进行充分的风险评估,重点考虑新增品种中不同成分的特性与预定用途等因素对现有品种可能产生的影响; 对于细胞毒性药物和非细胞毒性药物的共线生产,应从厂房设施、设备、工器具及风险控制措施方面防止污染与交叉污染的风险。
建议清洁验证时考虑产品的溶解性及设备材质选择适宜的清洁溶剂; 对残留检测分析方法进行验证,以保证检测结果的可靠性; 取样点应包括最难清洁点; 检测项目及限度应基于毒理试验数据或毒理学文献资料确定标准; 还要综合考虑阶段性生产的最长时间和最大批次数量,以作为清洁验证的评价依据。
5.5 环境监测方案的针对性
企业应基于风险评估的原则,根据药品特点和生产工序的洁净级别,针对性地制定环境监测方案,包括取样量、取样频次、取样位置等。如无菌工艺生产的B+A洁净区,在关键区域需有悬浮粒子、浮游菌、
沉降菌的监测点,需确保所有的无菌操作均进行了动态环境监测,无菌操作期间需关注在线悬浮粒子的报警情况,遇到报警需及时处理和调查。科学的环境监测方案有利于企业建立持续稳定的洁净环境,确保产品质量,降低污染的风险。
综上,本研究对20号公告涉及的化学仿制药注射剂品种注册检查中发现的问题进行了分析并提出应对策略。对于药品监管部门,建议对注射剂生产企业的薄弱环节进行重点监管。对于注射剂药品生产企业,应严格按照相关法规要求,结合品种生产工艺,
进一步提高无菌保证能力,全面提高我国化学仿制药注射剂的质量,充分保证药品的安全、有效和质量可控。
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: