三个指南对照:
|
机构 |
|||||||||||||||||
|
标题 |
化学仿制药透皮贴剂药学研究技术指导原则(试行) |
Transdermal and Topical Delivery Systems - Product
Development and Quality Considerations Guidance for Industry |
Quality of transdermal patches Adopted guideline |
||||||||||||||
|
时间 |
2020年12月 |
November 2019 |
Date for coming into effect - 17 June 2015 |
||||||||||||||
|
目录 |
一、概述 二、透皮贴剂仿制药药学研究的整体思路与要求 三、处方与制备工艺研究 (一)处方 1.原料药 2.辅料与材料 3.标识 (二)工艺 1.典型工艺步骤 2.工艺研究 3.批量 四、质量与特性研究 (一)一般要求 (二)特性质量研究 1.体外释放 2.体外透皮 3.体外黏附性能 4.药物残留 5.制剂中原料药热力学稳定性 6.黏合剂杂质 7.热效应 8.基质结构的微观评估 五、稳定性研究 六、名词解释 七、参考文献 |
I. INTRODUCTION II. BACKGROUND A. General B. Regulatory
Status III. TDS PRODUCT DEVELOPMENT A. Quality
Target Product Profile B. Critical
Quality Attributes 1. TDS
Product 2. Drug
Substance 3. Excipients
and Components 4. Identifying
Labeling C. Product
and Process Development IV. INFORMATION TO BE SUBMITTED IN AN APPLICATION A.
Pharmaceutical Development 1. Batch
Formula 2.
Expectations for Registration/Exhibit Batches 3. Product
Characterization Studies 4. Proposed
Manufacturing Changes B.
Manufacture C. Control of
TDS Product D. Additional
Stability Studies V. SPECIAL TOPICS A. Product
Adhesion Considerations B. Product
Storage and Disposal – Labeling Considerations |
Executive summary 1. Introduction (background) 2. Scope 3. Legal basis 4. New applications 4.1.
Description and composition of the drug product 4.2.
Pharmaceutical development 4.2.1.
Therapeutic objectives and principle of the delivery system 4.2.2. Active
substance 4.2.3.
Excipients 4.2.4.
Formulation development 4.2.5.
Stability programme development 4.2.6. In
vitro and in vivo drug product performance 4.2.6.1. In
vitro drug release / dissolution 4.2.6.2. In
vitro skin permeation studies 4.2.6.3.
Adhesive properties 4.2.6.3.1. In
vitro adhesion tests 4.2.6.3.2. In
vivo adhesion studies 4.2.6.4.
Pharmacokinetic studies 4.2.6.5.
Product batches used in clinical studies 4.2.7.
Manufacturing process development 4.2.8.
Container closure system 4.2.9.
Administration 4.3.
Manufacture 4.4. Control
of excipients, including layers and liners 4.5. Drug
product specifications 4.6. Control
strategy 5. Requirements to support a generic or abridged
application 5.1. General
remarks 5.2.
Development pharmaceutics 5.3.
Comparative quality and clinical data requirements 5.3.1.
Quality 5.3.2.
Clinical 6. Variations applications Definitions References Annex 1 (In vitro permeation studies) |
||||||||||||||
|
定义和范围 |
透皮贴剂(Transdermal Patch)系指用于完整皮肤表面能将药物输送透过皮肤进入血液循环系统起全身作用的贴剂。 本指导原则主要针对透皮贴剂化学仿制药。 |
Transdermal delivery systems are designed to deliver an active
ingredient (drug substance) across the skin and into systemic circulation,
while topical delivery systems are designed to deliver the active
ingredient to local tissue. This guidance provides recommendations to applicants
and manufacturers of transdermal and topical delivery systems (TDS)
regarding the pharmaceutical development and quality information to include
in new drug applications (NDAs) and abbreviated new drug applications
(ANDAs). |
A transdermal patch, which may also be
considered a Transdermal Drug Delivery System (TDDS), is defined as a
flexible, multi-layered, pharmaceutical single dose preparation of varying
size containing one or more active substances to be applied to the intact
skin for systemic absorption. This guideline considers the general requirements
concerning the development and quality of a transdermal patch for all new
marketing authorisation applications and variations. In addition, specific
guidance is provided concerning the data requirements to support generic or
abridged applications. |
||||||||||||||
|
全身作用 仿制药 |
全身作用、局部作用 新药、仿制药 |
全身作用 新药、仿制药 |
|||||||||||||||
|
分类和结构 |
透皮贴剂按照含有活性物质的支撑层的结构特点通常可分为骨架型(Matrix Type)和储库型(Reservoir Type)。 骨架型贴剂通常由背衬层(Backing Membrane)、含有活性物质的支撑层(Drug-in-Adhesive Matrix)、黏合层(Contact Adhesive)、保护层(Release Liner)等组成。 储库型贴剂通常由含药液态或半固态凝胶用热封区域截留在背衬层和控释材料之间制成。 |
Matrix type TDS contain one or more active ingredients dissolved or
partially suspended in a mixture of various components, including adhesives,
penetration enhancers, softeners, and preservatives, and are typically
manufactured using solvent, hydrogel, or hot melt-based practices. 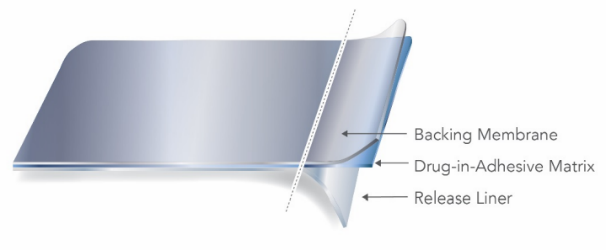 Reservoir type TDS similarly contain a variety of components in liquid
or semi-solid form; however, reservoir type TDS utilize a heat-sealed area to
entrap the active gel between the backing membrane and a microporous
membrane. 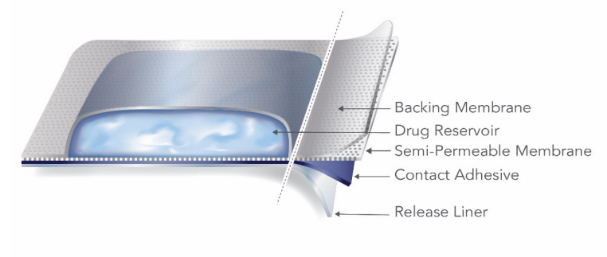 |
A transdermal patch includes a backing sheet,
impermeable to the active substance and normally impermeable to water. The
releasing surface of the patch is covered by a protective liner to be removed
before applying the patch to the skin. Alternatively, absorption may be limited by
incorporating or dissolving the active substance in a (semi solid) reservoir,
with a membrane to control the release and the diffusion of the active
substance(s) from the patch. |
||||||||||||||
|
制剂质量考虑 |
透皮贴剂的质量研究一般应包括但不限于以下研究:性状、鉴别、含量、单位剂量均匀性、有关物质、剥离强度、保护层剥离力、初黏力、持黏力、冷流、体外释放、原料药析晶、包装完整性、残留溶剂、微生物限度(如适用)、透皮促进剂含量/抗氧剂含量/含水量(如适用)等。 |
Quality Target Product Profile:
CQAs typically include appearance (such as lack of
visible crystals), dimensions, uniformity of dosage units, assay, permeation
enhancer content, impurities and degradants, in vitro drug release profile,
preservative/antioxidant content (if present), peel adhesion, tack, release
liner peel strength, shear strength, cold flow, residual solvents, residual
monomers, microbial limits, and package integrity. |
The development of the drug product should be
described with respect to the defined quality target product profile,
employing suitable tests to characterise and control the critical quality
attributes, including adhesion properties, factors affecting ease of
administration and duration of use, and product performance (dissolution, in
vitro drug release, in vitro skin permeation). In terms of quality in relation to efficacy: · The active substance content, formulation,
patch size and thickness should be justified by a sound rationale, in vitro
quality testing and clinical evidence, described by a clear narrative of
product development. In terms of quality in relation to safety: · Those quality elements that may influence
the safety of the drug product such as material specifications,
qualification, identification and control of residual solvents and impurities
should be discussed. The risks of dose dumping, leakage from reservoir,
residuals and product residues should be discussed. Cross reference to
relevant non-clinical or clinical data should be given. In terms of quality with respect to the
administration and use: · The adhesive properties of the drug
product should be fully discussed and characterised, by both in vitro and in
vivo testing. The adhesion / cohesion balance should be considered, with
respect to minimisation of cold flow (formation of a “dark ring” around the
transdermal patch in use), satisfactory elasticity and the avoidance of
detachment, or edge lifting throughout use and skin damage following removal. |
||||||||||||||
|
原料药质量考虑 |
应对可能影响透皮贴剂性能及生产可行性的原料药的理化及生物特性进行研究,特别是影响递送速率的性质,如分子量、熔点、分配系数、pKa、溶解性能和pH值等。 原料药的其他特性,如粒度分布、晶型与晶型稳定性等,应根据产品性能进行评估和论证,选择其特征指标进行研究。 |
Selection of a drug substance should be justified
based on the physicochemical and biological properties of the drug substance
that can influence the performance of the TDS product and its
manufacturability. In particular, properties that influence the rate of
delivery, such as molecular weight, melting point, partition coefficient,
pKa, aqueous solubility, and pH, should be considered. Other characteristics
of the drug substance such as particle size, crystal form, and polymorphism
should be evaluated and justified in terms of product performance. |
Active substance physicochemical and biological
properties that determine the capability and / or influence the rate and
extent of transdermal delivery and the manufacturability and stability of the
drug product should be identified and discussed. Such properties include
molecular weight, partition coefficient, melting point (boiling point if
applicable), pKa, solubility and pH effects, as well as physical properties,
such as particle size and polymorphism, if the active substance is present in
the solid state in the drug product. The target physical state of the drug substance, e.g.
solute, suspension, and the degree of saturation or supersaturation are
critical quality attributes and should be justified in terms of product
efficacy and safety, supported by evidence of the means by which the target
state is achieved during manufacture and its stability during storage. The risks of precipitation / particle growth / change
in crystal habit, or other active substance characteristics likely to affect
the thermodynamic activity, arising from changes in temperature and on
storage should be assessed and appropriate tests included in the stability
studies. These properties could be inter-related and might need to be
considered in combination. |
||||||||||||||
|
辅料质量考虑 |
透皮贴剂所使用的辅料与材料可能包括各种黏合剂、透皮促进剂、增溶剂、增塑剂、增黏剂、抗氧剂、稳定剂、交联剂、结晶抑制剂、控释膜、背衬材料、保护层等。 对于黏合剂,研究者应根据其用途考虑以下属性:分子量、多分散性、光谱特性、热力学特性、特性或复合黏度、残余单体与二聚体、残留杂质(如催化剂和引发剂)、残留溶剂、重金属等。其它需要考虑的属性包括黏弹性能,如弹性模量G'、黏性模量G,蠕变柔量J等。此外,应对黏合剂可能影响终产品质量(如体外释放、体外透皮和体外黏附性能等)的功能性相关指标进行研究。 对于膜性材料,应根据其不同用途进行相关研究。如控释膜、背衬材料、保护层等应对外观、柔韧性、抗拉强度、孔隙率、密封性、化学惰性等特性进行研究。 而对于控释膜型透皮贴剂,还应研究控释膜的适用性与性能。必要时,还应对膜性材料的提取物和浸出物进行研究。 对生产过程中使用但最终去除的物料(如临时膜材、溶剂等)进行必要的研究,评估上述物料组分转移并残留至终产品中所导致的质量及安全性风险。 |
Excipients and components used in TDS can include
various adhesives, permeation enhancers, rate controlling or non-rate
controlling membranes, solubilizers, plasticizers/softeners, or tackifiers,
all of which can influence the quality and performance attributes of TDS. For example, when qualifying the adhesives in a TDS
product, an applicant should consider the following attributes: · For adhesive polymer(s) as raw
material(s): molecular weight, polydispersity, spectroscopic analysis (e.g.,
infrared radiation (IR) absorption), thermal analysis, intrinsic or complex
viscosity, and measurement of residual monomers, dimers, solvents, heavy
metals, catalysts, and initiators. · For adhesive as a laminate (in the absence
of the active ingredient and other excipients): residual solvents, peel,
tack, shear, and adhesion. · For adhesive in the final product (along
with drug substance and other excipients and components): identification,
residual monomers, dimers, and solvents; impurities; loss on drying; and
uniformity. Other properties to be considered include the viscoelastic
properties (such as elastic modulus (G’), viscous modulus (G”), and creep
compliance (J)), and functional properties including, but not limited to,
peel, shear, adhesion, tack, in vitro drug release, and in vitro drug
permeation. |
The relevant characteristics of the layers (backing
layer, the release liner, rate controlling membranes and adhesive liner),
such as appearance, flexibility, tensile strength, porosity, occlusion and
chemical inertness, and the other excipients should be discussed. This
information can be used, as appropriate, to justify the choice and quality
attributes of the excipients, their specification and safety (3.2.P.4.4 and
3.2.P4.6) and to support the drug product specification (3.2.P.5.6). The composition and relevant characteristics of
excipient mixes, e.g. adhesive solutions or suspensions, should be provided
and characterised, including viscoelastic properties, if appropriate.
Processing aids, including temporary lamination layers and solvents employed
during manufacture, which are subsequently removed, should be identified and
described. |
||||||||||||||
|
包装系统质量考虑 |
|
|
This should include the choice of materials,
protection from moisture, oxygen and light, drug product compatibility and
safety. The primary package should normally contain only a
single transdermal patch. The backing layer and release liner should not be
considered a part of the container closure system. Appropriate tests should be included in the stability
study protocol to ensure that the suitability of the container closure system
is satisfactorily assessed throughout shelf-life. |
||||||||||||||
|
标签要求及质量考虑 |
标签标识一般印在透皮贴剂的背衬层上,至少应包括产品名称和规格。 对于管制类药物,应根据监管要求,确保在贴剂的全生命周期均具有足够的对比度和辨识度,可采用机械模拟试验(如摩擦等)与化学模拟试验(如喷淋、洗涤剂清洗等)来考察标识持久性。 应对标签标识的印刷材料与透皮贴剂之间的相互作用进行研究,以评估其对透皮贴剂的质量及安全性的影响。 |
Applicants are encouraged to incorporate a
representative label early in development to assure the labeling process or
inks utilized for printing do not interact with the TDS product, and to
properly assess inks during extractable and leachable studies. The
identifying label is typically placed on the backing membrane of TDS and
should, at minimum, include the product name and strength. |
The SmPC, package leaflet and labelling should fully
address the correct administration of the transdermal patch and include any
necessary warnings for the safe use of the drug product. |
||||||||||||||
|
生产工艺及过程控制 |
1.典型工艺步骤 透皮贴剂典型的生产步骤/单元操作通常包括但不限于:混合、涂布干燥、复合层压、分切、印刷、裁切和装袋。 2.工艺研究 透皮贴剂工艺较复杂,应加强中间体的研究和控制,应对可能影响产品关键质量属性的物料特性、工艺步骤及工艺参数进行研究。 混合工艺可能影响产品的含量、原辅料稳定性、含量均匀度、微观形貌和黏合剂的物理性能等。 需考察的工艺参数通常有物料加入顺序、混合速度和时间、温度、再分散或再循环条件、脱气条件等,需考察的物料属性通常有原料药粒度、晶型、物料的流变特性、溶剂型物料中固形物含量百分比等。 涂布干燥工艺可能影响产品的含量、含量均匀度、微观形貌、药物释放、稳定性、残留溶剂、残留的黏合剂杂质以及黏合剂基质的物理性质等。需考察的工艺参数通常有生产线速度、泵或螺杆速度、区域温度、空气流速、干燥空气的温湿度等。 需考察的物料属性通常有涂布混合物的流变性、均匀性以及涂布混合物中的溶剂和黏合剂杂质含量等。此外,还应对为补偿干燥期间的挥发而导致的原辅料过量投料进行研究。 应当对单元操作之间任何中间体的存放时间及条件进行考察。 |
Typical TDS manufacturing steps/unit operations are
listed below (a non-exhaustive list). For processes that incorporate these
steps, the applicant should describe how each operation and associated
controls were developed, addressing the considerations below, specifically,
the CQAs that may be impacted by the operation, and the relevant process
parameters and material attributes that may impact the output of each operation: · Mixing: Mixing operations produce bulk
mixtures for the coating step. Mixing can impact CQAs such as assay,
stability of drug substance and/or excipients, content uniformity,
microscopic appearance, and physical properties of the adhesive. The control
strategy should address the impact of equipment design, order of material
addition, and process parameters (such as mixing speeds, mixing times,
temperatures, redispersion or recirculation conditions, and deaeration
conditions) on CQAs, and should be justified, as necessary, based on
development studies. CMAs that can impact mixing include drug substance
particle size, polymorphic form, raw material rheological attributes, and
percent solids for materials supplied in solvent-based mixtures. · Coating is the application of a mixture to
a substrate. Depending on the equipment used, coating can impact CQAs such as
content uniformity and microscopic appearance. Though CPPs are equipment
dependent, firms should demonstrate that the control strategy (e.g., process
parameters to be controlled) is adequate to ensure content uniformity and
microscopic appearance for the full duration of the coating operation. CMAs
that can impact coating include the rheology of the bulk mixture and
within-roll uniformity of the substrate to be coated. · Drying involves the removal of solvent
from the mixture following the coating process. This process step can impact
CQAs such as assay, permeation enhancer content, antioxidant content, water
content (for hydrogels), content uniformity, microscopic appearance, drug
release, product stability, residual solvents, residual adhesive impurities,
and physical properties of the adhesive matrix. Therefore, CPPs for drying
that may need to be considered during process development include line speed,
the pump or screw speed, zone temperatures, air flow rates, temperature of
the drying air, and humidity of the drying air. Process development should
also consider the CMAs that can impact drying such as solvent and adhesive
impurity content in the bulk mixture. Applicants should also provide data to
justify any drug substance overage or excipient excess that may be needed to
compensate for any evaporation during drying. · Lamination involves the combining of
multiple layers of a given transdermal system design into a single common
laminate. Applicants should provide development data for corona treatments if
such a process is used to bond the adhesive to a backing film or rate-controlling
membrane. · Slitting and Printing: The bulk product is
typically slit longitudinally into narrower rolls of laminate for further
processing. Slitting and printing are typically low risk steps; however, if
certain aspects of the printing processes, e.g., excessive penetration depth
or heat input, can adversely affect product quality, then printing processes
should be characterized and controlled. · Converting and pouching: Converting and
pouching typically involve cutting a continuous laminate into individual
units and sealing the unit in a heat-sealed pouch. CQAs affected by these
processes include usability of the product (e.g., the ability to remove a
release liner) and pouch integrity. Common CPPs for these steps include heat
sealing temperatures and dwell times. · Curing: Some TDS have processing steps to
complete a curing reaction after drying or pouching. Curing time and curing
conditions are common CPPs for this step. Curing should be completed before
batch release testing if curing could impact test results. · Hold times: Hold times must be defined and
justified for in-process materials held between unit operations. Applicants
should use a risk-based approach to determine which CQAs to monitor during
hold time studies. · Other considerations: Tubing and other
product-contact equipment must be qualified as non-reactive, non-additive,
and non-absorptive. The selection of the tubing and certain
product-contacting equipment should be risk-based, i.e., dependent on the
duration of contact, process temperature, solvent system, material
considerations, clearance of leachables during manufacturing, and clinical
use considerations. In-process controls (IPCs) for TDS are an integral
part of the control strategy. The description of the proposed IPCs should
address the following: · At the mixing stage, IPCs can provide
assurance of assay, viscosity, uniformity, and pH for aqueous mixtures. If
multiple samples are taken from a dispersed mixture, applicants should
specify the mean, range for individual samples, and percent relative standard
deviation. · IPCs for coating, drying, and lamination
can provide assurance of uniformity across the laminate and throughout the
run. · For converting and pouching, IPCs can
provide assurance of pouch integrity, product placement within the pouch, and
product appearance. An automated system can perform in-process checks for
product appearance in lieu of human operators if the automated system is
demonstrated to be suitable for the intended task(s). |
The following non exhaustive list should be
discussed: · The preparation and homogeneity of the
bulk drug containing and if applicable the bulk non-drug containing adhesive
masses; · The coating process, including those
parameters that control the layer thickness; · Drying, curing and where applicable the
removal of residual solvents, including water for aqueous based blends; · Laminations steps; · The storage and handling of intermediate
rolls; · Roll conversion to transdermal patches;
and · Primary packing. |
||||||||||||||
|
制剂的规格 |
目前国内已上市透皮贴剂的规格有载药量、载药量/贴剂面积、递送速率等多种表达方式,而在欧美国家目前通常以递送速率,即递送量/释放时间(例如,**mg/天或**mg/h 或**mg/24h)表示,该递送速率可源于 PK 数据或残留药物分析数据。 |
The strength of a transdermal system should be
expressed as a rate (e.g., XX mg/day), whereas the strength of a topical
system should be expressed as a percent total drug load. For transdermal
systems, the strength can be derived from and supported by either PK data or
by residual drug analysis performed on used transdermal systems. The first
approach involves the derivation of a clearance (Cl) value from absolute
bioavailability of the drug and multiplying that by the concentration (Css)
at the steady state. The second approach involves the measurement of the
amount of drug left in the transdermal systems at the end of the wear period
and dividing the “consumed amount” by the wear period. |
Strength, as the mean dose delivered per unit time,
normally mass delivered in vivo per 24 hours. |
||||||||||||||
|
产品特性研究-原料药热力学稳定性 |
应确认原料药的热力学稳定性,评估生产和储存过程中析晶、沉淀或成盐的风险,以及对产品性能的影响。析晶研究可选用显微镜和光度法,以及 DSC、XRD 等分析手段。 |
To confirm thermodynamic stability of the drug
substance, the risk of precipitation or salt formation during manufacturing
and storage should be evaluated. If there is an equilibrium between different
salt forms, the kinetics to reach this equilibrium should be thoroughly
characterized. The impact of this equilibrium on TDS performance should be
evaluated with relevant in vitro drug release, permeation, and/or clinical
data. |
|
||||||||||||||
|
产品特性研究-药物残留 |
透皮贴剂中药物的含量通常高于使用过程中的递送剂量以达到临床有效给药率。由于活性成分的浓度可能接近于其饱和极限,产品在贮存过程中存在药物结晶的风险,并对产品的质量和疗效产生潜在不良作用。此外,给药后透皮贴剂中残留的药物对患者、他人和环境都存在风险。 应对透皮贴剂的药物残留特性进行研究,并在临床阶段对残留量进行实际研究,而不应仅根据理论计算或文献数据评估。仿制药的药物残留量不应超过参比制剂,否则应对其合理性进行说明。 |
To provide a robust analysis of the residual drug, we
recommend the following: 1. Data should be based on analysis of the used TDS
and not on a theoretical calculation. 2. The amount of drug left on the skin surface should
be assessed. Any drug that may have been transferred to packaging or other
components of the TDS during storage or use should be accounted for in an
attempt to perform a mass balance. 3. Tape or overlays should not be used in studies
where the TDS is used to calculate residual drug. 4. TDS adhesion assessments should be conducted over
the entire period of wear to determine whether the TDS diffusional surface
area remains in full contact with the skin during the entire period of the
study. 5. A control study should be performed to provide an
estimate of drug load, rather than simply using the expressed label claim.
This study should include analysis of a minimum of three unused products from
the same lot of product used in the study. 6. Sample storage conditions before and after
application of the TDS on the skin should be validated. Photostability and
thermal stability of the active ingredient(s) in the TDS should also be
considered when selecting the appropriate storage conditions. 7. Appropriately sensitive and valid analytical
methods should be used to assay the residual drug content for the purpose of
calculating drug depletion and delivery. When estimating the amount of
residual drug in the TDS, a drug extraction method with a target extraction
efficiency close to 100 percent should be utilized to minimize error. |
|
||||||||||||||
|
产品特性研究-体外释放试验 |
体外释放试验(IVRT)是评估药物从透皮贴剂释放的速率和程度,是质量研究及稳定性考察中的重要指标。体外释放度可以载药量百分比表示,也可以(单位时间内)单位面积的药物释放量表示。 透皮贴剂体外释放度研究方法主要有桨碟法和转筒法,在中国药典、美国药典(USP)和欧洲药典(EP)中均有收载。此外,美国药典(USP)还收载了往复支架法,日本药局方(JP)收载了纵向扩散池法。 在建立体外释放度考察方法时,应对介质、pH 值、装置、转速等测定装置和测定条件进行筛选和优化,最终选择区分力适宜的测试条件,释放度方法需进行充分的方法学验证。 为了解产品的释放特性,通常应选取足够多的取样测试点,以绘制完整的释放曲线(包括上升曲线及达到平台的阶段)。前期取样点的时间间隔应较短,后期取样点时间间隔可相对延长,直至 80%以上的药物释放或达到平台期(每 2 小时取样 1 次,连续三个时间点药物释放无增加),整体考察时间可视制剂释放时间长短而定,一般不宜短于药物作用时间。 在释放曲线研究的基础上,释放度取样点应选取初始、中间和最终阶段每个阶段至少一个点,以载药量百分比表示为例,在任何时间点所允许的释放变化量应不超过限度平均值±10%,除非有临床批次证明其合理性。各点的释放度限度平均值应基于自制品的临床批次、注册/申报批和商业批(如有)数据的统计评估。放行和货架期的限度值应相同,除非有临床批次证明其合理性。 |
In vitro drug release testing of TDS products is
typically performed using specific, qualified apparatus such as: Paddle over
Disk, Cylinder, or Reciprocating Holder. The NDA or ANDA submission for the TDS product should
include a method development and validation report with complete
information/data supporting the proposed drug release method and acceptance
criteria. Sufficient detail and data should be included in the
method development and validation report so the adequacy of the method for
batch release and stability testing can be properly assessed. Examples of
parameters to evaluate during method development include selection of USP
apparatus/other equipment, drug release medium, rotation or agitation speed,
temperature, pH, sink conditions, use of a surfactant, and other technical
aspects of the test. An in vitro drug release method should be simple,
reliable, reproducible, discriminating, and robust. Applicants should strive
to develop a method that releases as much drug as possible. The validation section of the report should include
complete information/data regarding: i) the discriminating ability of the
selected method, ii) the validation of the drug release methodology, and iii)
the validation/verification of the analytical method selected to assay the
drug release samples. The selected method should be able to differentiate the
release profiles of TDS that are intentionally manufactured with meaningful
variations in critical process parameters and formulation components. Validation
data should demonstrate the range and sensitivity of the method for
proportional drug release across different strengths of the TDS. In addition,
validation data should demonstrate reproducibility of the method for drug
release across different runs of the same batch and its robustness, i.e., its
capacity to remain unaffected by changes in receptor medium temperature,
paddle rate, and other method parameters. The acceptance criteria for the in vitro drug release
test should be based on the proposed TDS product batch release data,
including data from bio-batches (e.g., BE, PK, Clinical),
registration/exhibit batches, and commercial batches (if available). To set
the acceptance criteria for the in vitro drug release test, a complete drug
release profile should be established by collecting data until there is no
increase in drug release over three consecutive time points (sampling every 2
hours). The drug release profile of TDS products typically encompasses
initial, middle, and terminal phases; thus, for setting the acceptance
criteria, there should be at least one sampling time point covering each
phase. The drug release data should be reported as the cumulative percent of
drug being released with time. The acceptance criteria range for each
specific timepoint should be based on the mean percentage value of drug
released ± 10 percent using the drug release data generated at these times.
The percentage should be determined based on the TDS product’s label claim.
If less than 100 percent drug is released, but no drug increase is observed
over three consecutive sampling timepoints (i.e., incomplete drug release),
the amount of drug reached at the plateau should be considered 100 percent
for the purposes of estimating the percent of drug release over time. Wider acceptance criteria range for the drug release
test may be acceptable if they are supported by an approved in-vitro in-vivo
correlation model. |
An in vitro release test evaluates the rate and
extent of release of an active substance from a transdermal patch. Although
the test may not model in vivo performance, it is a critical quality
attribute to be specified in the finished product release and shelf life
specification. The methods described in Ph. Eur. monograph for
Transdermal Patches should be followed i.e. a dissolution test or a release
test using an appropriate, non rate-limiting membrane. If appropriate,
alternative methods, with improved discriminative power compared to the
compendial methods, may be employed. The test itself and / or sample preparation should
not damage or otherwise alter the performance of the transdermal patch. Any
special requirements for sample preparation should be discussed. It may be
possible to test only a defined sample area of patch which is applicable to
all strengths, if it is shown that sample preparation has no impact on drug
release / dissolution. If the size of the patch is too large to be inserted
into standard dissolution testing apparatus or if sink conditions cannot be
achieved using entire patches, suitability of testing specimens might be
inferred from dose proportionality studies for samples of different size. The in vitro drug release / dissolution profile of
the active substance from the drug product should be characterised and
established from clinical batches for which satisfactory efficacy has been
demonstrated. These should be used to support the in vitro drug release /
dissolution limits in the drug product specification (3.2.P.5.6), and so
provide an assurance that future production batches are of similar quality to
the pivotal clinical batches. Satisfactory evidence of discrimination should be
provided, with respect to: · Critical manufacturing variables; · Excipient and active substance critical
quality attributes; and · The stability indicating power of the
method. A summary of the development of the dissolution test
should be provided, where the transdermal patch is tested under various
conditions (media, pH, apparatus, agitation, etc.). Testing conditions
providing the most suitable discrimination should be chosen. In case of media
with a low buffering capacity, the pH should be controlled during the
dissolution test to avoid influence of the dissolved active ingredient and/or
excipients on the dissolution conditions during the test period. The test period should be sufficient to achieve
complete drug release, unless justified. For the release / dissolution profile, the number of
sampling time points should be sufficient to obtain meaningful profiles, with
more frequent sampling during the period of greatest change. At least 3 sampling time points are recommended to
give a sharper and more differentiated profile. An early time point to exclude dose dumping and/or to
characterise a loading/initial dose (typically 20 to 30% dissolved), at least
one point to ensure compliance with the shape of the dissolution profile
(around 50% dissolved) and one to ensure that the majority of the active
substance has been released (generally more than 85% dissolved i.e. Q=80%). For most matrix type patches earlier sampling times
(between 0 to 1 hour) were found to be more discriminative, i.e. quality
indicating than later time points, when already up to 50% of active substance
is released from the patch. Changes in formulation or manufacturing
parameters are more likely to be detected within the first hour of in vitro
dissolution testing if the specification ranges are set in accordance to the
requirements listed below. For the dissolution profiles, the value to be
reported at each time point should be the quantity of active substance
released in mass units (mg or μg) per surface area. The quantity of active
substance should also be reported as a % of the total. In addition, the first derivative of this profile
should also be reported, to allow assessment of the change in the rate of
release over time i.e. the value to be reported at each time point should be
the quantity of active substance released per surface area, per time. For transdermal patch products showing an in vitro
zero order release (e.g., which may be seen in those patches with a
rate-controlling membrane) a specification of the dissolution rate at a given
time point may be more appropriate than the cumulative amount dissolved at a
given time point. The number of samples used to characterise the
dissolution profiles should normally be a minimum of 12 units per batch (for
routine release, a minimum of 6 units would be accepted). Dissolution profile data should be provided in
tabular and graphical formats, with a measure of variability between units
e.g., 95 % confidence interval, range, or other justified statistical
approach. The dissolution profiles should be discussed taking
into account the type of transdermal patch. Dissolution limits should be data driven and fully
justified. They should be characteristic of production capability and in line
with clinical batches for which satisfactory efficacy has been demonstrated. Normally, the permitted range in mean release at any
given time point should not exceed a total numerical difference of ±10% of
the labelled content of active substance (i.e. a total variability of 20%: a
requirement of 50±10% thus means an acceptable range from 40-60%), unless a
wider range is supported by a bioequivalence study. Wider limits may be accepted only if satisfactorily
explained and justified on quality grounds and supported by a bioequivalence
study. Release and shelf life limits should normally be the
same, unless the reasons for the differences are satisfactorily explained on
quality grounds and justified by reference to clinical batches. Tighter
limits at release should be set to ensure that the product will remain within
the shelf life specification. |
||||||||||||||
|
产品特性研究-体外透皮试验 |
体外透皮试验(IVPT)是为了模拟药品在生理条件下的透皮过程,以部分地反映药品的质量与临床治疗的有效性。应在体外透皮试验方法系统的研究及验证基础上,对仿制药与参比制剂进行皮肤透过率的对比研究。 体外透皮试验目前主流方法为 Franz 扩散池法(Diffusion cells),也可采用流通池法(Flow Through Cell)或其他经过方法学研究证明可行的体外透皮试验方法。 在建立体外透皮试验方法时,应对接受介质、温度和转速、皮肤种类与部位以及皮肤完整性进行研究,选择具有适宜区分力的试验条件开展体外透皮试验研究。 |
In vitro permeation testing (IVPT) with the use of
excised human skin may be utilized to characterize the rate and extent of
transdermal or topical drug delivery, and the study protocols and results
should be described in the application. The following factors should be
considered during IVPT model development: · Selection of the diffusion apparatus and
the operating conditions like stirring rate or flow rate, as well as
temperature control to maintain the under-normal-conditions skin surface
temperature (32°C ±1°C) · Source of the skin, skin storage
conditions, choice of skin type (i.e., age range, sex , race, and consistent
anatomical region) and the skin preparation technique (e.g., full-thickness,
dermatomed, isolated epidermis) The IVPT protocol should specify the nominal skin
thickness and its range, details of the skin barrier integrity test, and any
occlusion of the product during the IVPT. Visual observations alone are not
sufficient to characterize the barrier integrity of the skin. Acceptable
barrier integrity tests may be based on tritiated water permeation,
trans-epidermal water loss (TEWL), or electrical impedance/conductance
measured across the skin. The test parameters and acceptance criteria used
for the skin barrier integrity test should be justified based on relevant
literature references or other information. The IVPT protocol should also include details about
the receptor solution, system equilibration, procedures for skin mounting and
application of the TDS, as well as any measures to secure the TDS on the skin
surface to prevent lifting. We recommend that an antimicrobial agent be
included in the receptor solution (e.g., ~0.1 percent sodium azide or ~0.01
percent gentamicin sulfate). The IVPT study report should include dose duration,
sampling duration, sampling time points, concentration of samples,
concentration of the antimicrobial component, and the empirical stability (at
relevant temperatures) and solubility of the active ingredient in the
receptor solution. The study report should also include the number of
individuals whose skin was evaluated (i.e., skin donors) and the number of
replicate skin sections per donor per treatment group. All treatment groups compared in an IVPT study should
be dosed on the skin samples from the same set of donors, with the same
number of replicates per donor per treatment group. These treatment groups
should also use the skin samples from the same anatomical site from all
donors, unless varying these parameters is essential to the design of the
study and the evaluation of the TDS. The study report should include the
equilibrated skin surface temperature prior to dose application, and the
ambient temperature and relative humidity in the laboratory, as well as the
extent of qualification of the sample analytical methods (e.g., HPLC). |
1. Introduction Percutaneous/dermal absorption describes the passage
of compounds across the skin: · Penetration which is the entry of a
substance into a particular layer or structure such as the entrance of a
compound into the stratum corneum; and · Permeation which is the penetration
through one layer into another, that is both functionally and structurally
different from the first layer. In vitro permeation studies are often used throughout
pharmaceutical development to evaluate the permeation of an active substance
from a transdermal patch. A major advantage of in vitro studies is the
possibility for controlling the conditions of the experiment and therefore
changes in permeation should only arise from changes in the transdermal patch
and/or the membrane used. In vitro permeation studies are not expected to
correlate with in vivo release, but the characterisation of the permeation
profile is considered a valuable measure of product quality and performance
and may reflect the thermodynamic activity of the active substance in the
product. Establishing the characteristic permeation profile of
the drug product, using a discriminative in vitro skin permeation method, can
be of value in change control during life cycle management. For the comparison of products, the need to perform
bioequivalence studies may, in certain cases, potentially be waived. However,
given that the product formulation may have a great impact on efficacy and
modify skin permeation, the products to be compared should have the same or
similar qualitative and quantitative excipient composition. In line with the requirements for in vitro drug
release / dissolution methods, satisfactory evidence of discrimination and a
summary of the development of the permeation test should be provided. It is recommended to use human skin from the torso
(breast, abdomen or back) or relevant to the site of clinical application.
However, if not possible, non-viable skin or skin from other species (such as
pig, rodent, guinea pig) can be used. In some cases, artificial/synthetic
membranes can be suitable. The choice of skin model used throughout the
development should be justified. In the case of a comparative test (development of a
generic transdermal patch or formulation comparison), it is necessary that
the products are compared with the same skin type. The skin type, preparation and storage are elements
that should be satisfactorily controlled, and that the skin samples for
experiment are not damaged by these processes and are of suitable quality. The integrity of the skin should be determined before
the experiment and shown to be satisfactory for the experiment to be valid. A variety of integrity tests is currently available,
e.g., measurement of Transepidermal Water Loss (TEWL), monitoring the
permeation of tritiated water or measurement of electrical resistance. The
suitability of integrity test for the proposed experiment and criteria for
acceptability should be fully discussed. To address the variability of skin, preparation and
skin integrity, a sufficient and justified number of replicates should be
included in the experiment. Typically, six replicates may be used, or more
for pivotal experiments. In vitro permeation studies are usually performed
using diffusion cells, with a skin diffusion area between 0.5-2 cm2.
Diffusion cells with larger skin diffusion areas should be used for
transdermal patches that cannot be cut to size, e.g., reservoir type patches.
Diffusion cells should be inert, robust and easy to handle. It is also
important that the diffusion cell provides easy sampling and replenishment of
the receptor phase and that it maintains the membrane integrity. The most
common diffusion cells are Franz-type (static) cells, which consist of two
chambers that can be side-by-side or upright separated by the skin. Flow
through cells can also be used and are particularly useful to maintain skin
viability. The receptor solution used should mimic the in vivo
conditions. One appropriate receptor medium for water soluble drugs is
aqueous buffer. Solubilising agents e.g., surfactants or hydro-alcoholic
media e.g., ethanol/water media, or protein e.g., bovine serum albumin can
also be used in the case of drugs that are less soluble in water, if
justified. The liquid in the receptor compartment has to be in contact with
the skin, i.e., it should be ensured that there are no air bubbles under the
skin. The receptor solution should not affect skin
integrity. Permeation of solubilising agents from the receptor
solution into the skin sample should be considered and avoided. The composition of the receptor medium should be
described and solubility studies to demonstrate that sink conditions can be
maintained throughout the experiment should be submitted. The active substance should be stable in the receptor
solution for the duration of the test and subsequent analysis. The receptor solution should provide solubility sink
conditions throughout the experiment and ensure that the permeation of the
active substance is not limited by the receptor medium. An acceptable sink
condition is one where the maximum concentration of the active substance in
the receptor solution achieved during the experiment does not exceed 10-30%
of its maximum solubility in the receptor solution. Sink conditions can be
maintained during the experiment in static cells by continuous replacement of
the receptor phase or by using a flow through system. For satisfactory permeation, satisfactory means
should be in place to ensure that the receptor medium is fully in contact
with all the exposed skin. The diffusion of the active substance through the
membrane is evaluated measuring the arrival of drug in the receptor
compartment by assay of sequentially collected samples of the receptor fluid. Aliquots of the receptor fluid can be analysed by a
validated HPLC or LC-MS for active substance content or by any other suitably
validated analytical technique. It is acknowledged that the variability in results
seen with in vitro skin permeation data is related to the variability in the
skin used. In addition, if these methods are poorly developed, without
satisfactory validation and / or poorly executed, then the results from
permeation studies can only be difficult to interpret or without merit or
meaning. Therefore, method development, optimisation and execution should
comply with known best practice, be satisfactorily validated, and subject to
appropriate data analysis and quality assurance principles. 2. Skin and sample preparation The following should be described and their
suitability discussed: · The type of skin (origin, species, part of
the body) should be stated; · The storage and transport of the skin
should be described and appropriately controlled; and · The preparation and treatment (thickness,
separation) of the skin should be described and justified. The integrity of the skin should be determined before
the experiment and shown to be satisfactory for the experiment to be valid. Sample preparation of the drug product transdermal
patch should be described and its suitability discussed. Normally, the patch
is carefully cut to size and applied to the skin in the donor chamber. In
case overlays are used, the effect of occlusion should be investigated. The test itself and / or sample preparation should
not damage or otherwise alter the performance of the transdermal patch. 3. Study design / study conditions The following study design is recommended for
permeation studies using ex vivo human skin. Any deviations from the proposed
test protocol should be fully justified. · Diffusion cell – Franz type or flow
through; · Receptor phase, to mimic in vivo
conditions that also provides active substance sink conditions, degassed,
e.g., in an ultrasound bath to prevent the build-up of air pockets; · The medium may be aqueous buffer and
include justified solubilising agents and / or protein; · Receptor phase should be continuously
agitated and remain in contact with the skin. The stirring speed should be
justified; · Temperature - the surface of the skin, in
the diffusion cell, is maintainable at a temperature close to the
physiological human skin temperature (32°C±1°C). The skin surface temperature
may be suitably verified prior to dose application using an infrared
thermometer; · Humidity – Extremes of relative humidity
in the laboratory should be avoided, i.e. above 70% RH and below 30% RH; · Human skin integrity should be tested at
the beginning of the experiment; · The suitability of integrity test e.g.,
(TEWL), permeation of tritiated water, electrical resistance or visual
inspection (but not accepted for pivotal studies) and criteria for acceptance
should be fully discussed; · Number of replicates – The choice of the
number of samples should be justified with regard to the scope of the
experiment and demonstrated to be statistically relevant; · Number of skin donors – at least 2
different donors; · Skin anatomical region –torso (breast,
abdomen or back) or relevant to the site of clinical application; · The number of time points should be
sufficient to satisfactory characterise the permeation profile. Minimum of 5
suitably timed receptor sampling time points and an early time point, based
on study requirements; · Study duration should be justified in
regards to the in-use administration. If the study duration is longer than 24
hours, it should also be shown that skin barrier function and integrity is
adequately maintained; and · Un-occluded conditions, in case overlays
are used the effect of occlusion should be investigated. 4. Method development and validation A summary of the method development and optimisation
should be provided. The most appropriate receptor medium for water
soluble drugs is aqueous buffer. Hydro-alcoholic medium or indeed any other
appropriate medium can be used in the case of drugs that are sparingly
soluble in water, provided that it is justified. Testing conditions providing
the most suitable discrimination should be selected. The composition of the
receptor phase should not influence the permeation of drugs, should ensure
sink conditions and should not alter the membrane. The method should be suitably discriminating. The
following should be considered: · It is discriminating between batches with
respect to critical manufacturing parameters that are known to have an impact
on the bioavailability of the product. · It is discriminating between products
formulated at different concentrations, and altered formulations (e.g., drug
content, permeation enhancer). · The stability indicating power of the
method should be assessed. The analytical methods for determining the content of
active substance in the receptor fluid and mass balance determinations should
be provided and validated according to ICH Q2(R1). The reference standards used during the validation
and study sample analysis should be obtained from an authentic and traceable
source. The method validation should also address the
variability of the method and the coefficient of variation established. For
artificial membranes, the coefficient of variation should be less than 10%,
for human skin a coefficient of variation greater than 10% can be accepted. 5. Data analysis Data from all diffusion cells should be reported and
the validity, variability and reproducibility of the results should be
discussed. The results should be subject to an analysis of variance. Outliers may be excluded from the statistical
analysis, if satisfactorily explained and justified. The plot of the
cumulative amount of drug permeated per unit area (mass/cm2) as function of
time should be presented. The slope of the curve represents the permeation
rate (flux) of the drug. The permeation profile should be supported with
tabulated data and statistical analysis. The permeation rate (flux) should be
calculated for each diffusion cell and the mean flux reported together with
the corresponding standard deviation (SD), coefficient of variation. A discussion, interpretation and conclusions of the
results of the experiment should be provided, supported, as necessary, by
appropriate scientific rationale. For the comparison of products, relevant permeation
parameters, e.g., flux, should be statistically compared. The 90% confidence
interval for the ratio of the two products should be determined and should be
contained within the ratio of 0.8 to 1.25 to support a claim of equivalence,
unless justified. The method should be based upon a null hypothesis of non
equivalence. 6. Quality system and study report It should be ensured that the performing laboratory
is qualified to perform the studies and that an effective quality system is
in place. This should include: · A declaration of compliance with a
suitable quality system, e.g., GMP (Directive 2003/94/EC); · The technical ability of the performing
laboratory and the validity of the method used should be assessed at regular
intervals, at least twice per year, by using reference compounds like
caffeine or benzoic acid. The recent results of such studies should be
provided; · The laboratory should be subject to
external audit e.g., by the Marketing Authorisation Applicant or a suitable
accreditation body; and · Audits certificates, if available. |
||||||||||||||
|
产品特性研究-可提取物和可浸出物 |
|
All TDS should be evaluated for potential compounds
that could be transferred from the product to the patient. This evaluation
should include assessments of extractables and leachables, consistent with
USP <1663> and <1664>. In the context of this guidance, extractable
impurities are chemical entities that can be drawn out of the backing
membrane, release liner, pouching material, printed ink, internal membranes,
and components other than the drug substance and adhesive matrix by a solvent
system. Additionally, an extraction study can detect compounds introduced
into the TDS from the manufacturing process, which can impact the final
impurity profile of the TDS product. In the context of this guidance,
leachables are chemical entities present in a packaged TDS because they
leached into the adhesive matrix (or where applicable, reservoir) under
normal conditions of storage or during accelerated stability studies. These
compounds may transfer from the adhesive matrix (or reservoir) to the patient
during use. i. Extractable Studies Extractable studies should be conducted early in the
pharmaceutical development process to understand the potential leachables
from components of the proposed commercial TDS. These studies should be
conducted on components such as backing membrane, release liner, rate
controlling or other internal membranes, ink and pouching. The testing
components should be extracted in a variety of solvents with a range of
polarities under vigorous laboratory extraction conditions to maximize the
levels of extractables and identify as many potential leachables as possible.
One of the extraction solvents used in the extractable studies should include
the solvent of the proposed commercial adhesive(s) platform or the known
residual solvents for the finished TDS. The choices of solvents used should
be justified. ii. Leachable Studies The conditions of the leachable studies should mimic
as closely as possible the “worst-case” clinical conditions of the skin
(e.g., sweating during rigorous exercise). The solvent/solution selection
(such as salt concentrations), temperature, level of agitation, duration of
exposure to the solvent, etc., selected for the studies should be justified.
The release liner should be removed from the system during the study to
adequately expose the adhesive layer to the biologically relevant solvent.
Applicants should conduct a multi-timepoint leachable analysis (e.g., 0, 6,
12, 24 months) to provide a comprehensive leachable profile and identify any
trends in leachables as these data could impact the shelf life of the
product. At the time of application submission, data should be submitted from
a leachable study performed on samples from multiple batches stored at a
minimum of 6 months under accelerated and long term conditions. We recommend
conducting leachable studies on the same three distinct laminates of TDS placed
on stability testing. |
|
||||||||||||||
|
产品特性研究-评估热效应 |
应考虑各种因素导致的体温升高对药物(特别是治疗窗较窄的药物)释放速率及透皮性能的影响,必要时进行相关研究,以评估仿制药与参比制剂的变化趋势是否一致。 |
Heat from external sources such as a heating blanket,
and potentially from a rise in internal body temperature due to strenuous
exercise or fever, may affect the rate of drug release from the TDS and the
absorption of drug into and through the skin. We recommend that applicants
study the impact of an elevated TDS/skin surface temperature on the delivery
profile of TDS relative to its delivery profile at a normal TDS/skin surface
temperature. For a TDS product to be submitted in an NDA, we
recommend that the heat effect studies be conducted as part of a clinical
study using the proposed commercial product. In designing the heat effect
studies, critical factors such as appropriate elevated test temperature(s),
heat exposure onset time(s), duration(s), and cycles (if any), as well as
mechanisms of heat exposure (e.g., heating lamp, heating pad, etc.) should be
identified. For a TDS product to be submitted in an ANDA, the
applicant should evaluate whether the test TDS, used under elevated
temperature conditions, increases drug delivery compared to the reference (R)
TDS. The ANDA applicant should provide the results of an IVPT study comparing
the drug delivery characteristics for the test TDS and the R TDS at normal
and elevated temperatures using skin from multiple individuals (donors), with
multiple replicate diffusion cells evaluated per donor, per treatment (test
versus R), and per temperature condition. An IVPT study with a sufficient
number of donors and replicates per donor per treatment per temperature
condition is recommended to obtain meaningful data. A study with fewer than
four donors and four replicates per donor per treatment per temperature may
be difficult to interpret. We recommend a parallel evaluation and comparison of
the test and R TDS under the following baseline and elevated temperature
conditions: 1. BASELINE: Both the test and R products should be
maintained at a TDS/skin surface temperature of 32 ±1°C for the entire study
duration. 2. ELEVATED TEMPERATURE: Both the test and R products
should be maintained at a TDS/skin surface temperature of 32 ±1°C until a
specified time, approximately when the peak flux is observed, and then
maintained at a TDS/skin surface temperature of 42 ±2°C for a period
thereafter, which may be the remainder of the study duration. It should not be assumed that a set temperature for a
circulating water bath will provide the target temperature at the TDS/skin
surface. The TDS/skin surface temperature should be directly measured using
an infrared thermometer or other temperature probe. The study duration for a
7- day wear TDS need not encompass the entire labeled duration of wear. It
may be adequate to perform an IVPT study for a 48 or 72 hour duration, if
that duration is sufficient to reach the peak drug delivery rate under baseline
conditions. Alternatively, an applicant may justify evaluating other
conditions or scenarios of exposure to elevated temperatures that represent
the worst-case scenario for a given TDS product or indicated patient
population. i. Microscopic Matrix Evaluation Due to complexities of many TDS formulations,
adhesive matrices often do not form true solutions, rather they manifest as
dispersions. If rearrangements of the dispersed-like system occur over time
within the matrix, they can possibly lead to lack of adhesion or changes in
drug delivery and release. As such, it is important to have a good
understanding of the TDS formulation, the way the drug substance and
excipients are dispersed within the adhesive matrix, and the tendency of the
matrix to change over time from product release through its expiry period.
Therefore, it is informative to assess surface and cross-sectional changes in
the TDS matrix throughout the shelf life of the developmental batches using
high-powered microscopy, elemental mapping, or other appropriate tools. These
tools may not be appropriate for every TDS; applicants should provide a
scientific justification for the tools used. These assessments will help
achieve comprehensive understanding of product and process, mitigate
quality-related risks, and assure that the TDS meets the requisite quality
attributes through its expiry period. |
|
||||||||||||||
|
产品特性研究-体外粘附性能 |
剥离强度:将透皮贴剂在规定压力下施用于标准基板,在指定的温度和时间下进行平衡,然后使用仪器将透皮贴剂从基材上剥离并记录相应的力。 保护层剥离力:用成品样品进行,在指定的温度和时间下进行平衡,然后使用仪器将保护层从透皮贴剂上剥离并记录相应的力。 初黏力:可采用滚球法、探针法或其他替代方法测定。 持黏力:可进行动态和静态测试。在动态测试期间,可将透皮贴剂以恒定的速率从测试面板上拉出。进行静态测试时,可通过悬挂砝码来测试透皮贴剂的承受剪切力。 冷流:冷流是透皮贴剂固有的特性,大小通常取决于药品处方、包装设计、贮存条件和时间。应重视处方的流变学研究,并采用定量和定性相结合的方法评估冷流。 黏附性能的限度范围应根据产品开发、稳定性考察及临床试验等多个产品批次的统计评估,确保各批次之间的黏附性能一致。 |
Using currently available methods, in vitro adhesion
testing does not correlate to in vivo adhesion, but in vitro adhesion testing
can be useful for quality control (QC) purposes. In vitro adhesion testing
should include peel adhesion, release liner removal, tack, and shear (dynamic
or static). The peel adhesion test measures the force required to
remove (peel away) a TDS that has been applied to a standard test panel
(e.g., polished stainless steel). The measurement of peel adhesion is
influenced by the test parameters such as dwell time, substrate (e.g.,
stainless steel, high density polyethylene (HDPE)), peel angle, and peel
speed. A release liner peel test measures the force required
to separate a TDS from its release liner. The measurement of release liner
peel is influenced by experimental parameters such as peel angle and peel
speed. The probe tack test measures the force required to
separate the test probe from the adhesive of the TDS. Tack measurement is
influenced by the test parameters such as the contact area, the contact
pressure, the time of contact (or dwell time), and rate of separation. There are two categories of shear testing, namely
dynamic and static. In the dynamic test, the TDS is pulled from a standard
test panel (e.g., polished stainless steel). Dwell time, speed, type of test
panel, mode of failure, and sample size are the typical test parameters
reported for the dynamic shear test. In the static shear test, the TDS sample
is applied to a test panel that is at an angle 2° from the vertical, and the
sample is subjected to a shearing force by a means of a given weight (e.g.,
1000 g) suspended from the TDS; the time required to detach a standard area
of the TDS from a stainless steel test panel under a standard load is
measured. Dwell time, weight used, type of test panel, mode of failure, and
sample size are the typical test parameters reported for the static shear
test. The time taken for the TDS sample to detach from the test panel is also
reported. Cold flow is the creeping or oozing of the adhesive
matrix beyond the perimeter of the backing membrane or through the release
liner slit. Cold flow may be present on the TDS, release liner, pouch, or
disposable films (sometimes termed slip sheets or protective films, such as a
film over the backing and a film over the release liner). Though a
quantitative method of assessing cold flow can provide a meaningful
measurement, it may not describe the difficulty in removing the TDS from the
pouch or the protective films from the TDS. The most accurate cold flow
assessment for TDS will likely come from a combination of product-specific
quantitative and qualitative methods. |
In vitro adhesive tests should characterise the
adhesion/cohesive properties of the transdermal patch. Although these tests
may not model in vivo adhesion, they are critical quality attributes to be
specified in the finished product release and shelf life specification. Tests to characterise adhesive properties may
comprise tests such as peel force tests (force required to remove the patch
from the release liner), adhesive strength tests (force required to remove
the patch from a defined surface) and tack tests (maximum force required to
break a bond formed under low pressure between the adhesive layer of the
patch and a stainless steel probe). Residue remaining on the release liner after peeling
from the patch and skin residues, following transdermal patch removal, should
also be addressed. The range and sufficiency of the in vitro tests used
to characterise the adhesive properties of the drug product should be
justified. A summary of their development should be provided to support any
justification, if necessary. The suitability and discriminatory power of the test
methods employed to characterise the adhesive properties of the drug product
need to be proven during product development, in particular with respect to: · Critical manufacturing variables; · Excipient and/or active substance critical
quality attributes; and · Stability indicating power of the method. The in vitro adhesive properties of the drug product
should thus be characterised, with the specifications limits for the
specified tests in accordance with the results obtained on clinical batches
for which satisfactory in vivo adhesive properties under product use have
been demonstrated and used to support their justification of the drug product
specification (3.2.P.5.6) (see also Section 4.2.9 Administration). Release and shelf life limits should be the same,
unless justified by reference to clinical batches. |
||||||||||||||
|
产品特性研究-体内粘附性能 |
|
In vivo adhesion studies provide the greatest
prediction of adhesion, a CQA, for a proposed commercial product. Applicants
should demonstrate that reasonable efforts were made to optimize adhesive
characteristics of the TDS. This optimization should balance properties such
as adhesiveness, cohesiveness, and stability to ensure a consistent and
uniform adhesion of its entire surface area to the skin for the entire
duration of wear. Applicants should develop a comprehensive strategy for
assessing the adhesive attributes of the TDS. In vivo adhesion studies are
necessary to demonstrate adequate adhesion of the TDS. Therefore, when
possible, such as in efficacy studies for an NDA, subject diaries describing
the actual in-use product adhesion performance should be used. This
information bolsters adhesion data collected from the studies described below
and in other guidances. Characterization of the adhesive properties of a TDS
should demonstrate that the labelled uses are substantiated. For example, if
the TDS is intended to be worn during bathing and showering, applicants
should demonstrate that the TDS will continue to adhere during and after such
incidental exposure to water. Product reinforcement, such as taping the edges
or use of overlays, or occluding the product from water during bathing should
not be permitted during the in vivo adhesion evaluation. We recommend that when assessing the adhesion of a
TDS, applicants use a 5-point numerical scale in which each score corresponds
to a specified range of adhered surface area of the TDS, as follows: 0 = ≥ 90% adhered (essentially no lift off the
skin) 1 = ≥ 75% to < 90% adhered (some edges only
lifting off the skin) 2 = ≥ 50% to < 75% adhered (less than half
of the TDS lifting off the skin) 3 = > 0% to < 50% adhered (not detached, but
more than half of the TDS lifting off the skin without falling off) 4 = 0% adhered (TDS detached; completely off) Additionally, the following information should be
collected: · At each time point when adhesion is
assessed on the above described 5-point scale, the scorer should also record
their actual percent adherence estimate (e.g., if the observer scores the
product as a two on the five point scale and estimates that the product
appears to be 60 percent adhered, a score of two and a 60 percent should be
recorded for that time point). · Photographic evidence showing the extent
of TDS adherence to the skin at each time point should be provided. |
Studies to investigate and establish the satisfactory
in vivo adhesive performance of the drug product should be undertaken. Since an in vivo adhesion study is pivotal for
approval, a feasibility or pilot study could be helpful in ensuring the
methods can be satisfactorily undertaken, producing result from which valid
conclusions can be made. The assessment should be undertaken throughout the
proposed period of use of the patch. This is because satisfactory adhesion
performance of the clinical batches used would be a requirement for any
clinical conclusions to be valid (see also Guideline on the Pharmacokinetic
and clinical evaluation of modified-release dosage forms). The clinical batches should be representative of the
product to be marketed (see Section 4.2.6.5 Product Batches used in Clinical
Studies). Reference to the in vivo adhesion studies described
in the clinical dossier should be provided. |
||||||||||||||
|
产品质量控制项目 |
应根据产品特点制订质量控制项目,除通用质量控制项目(如性状、鉴别、含量、单位剂量均匀性、有关物质)外,还应至少包括体外释放及黏附性相关的检查项。相关项目的可接受标准应结合参比制剂测定结果,依据自制品临床代表性批次质量研究数据与稳定性数据制定。 |
Typical CQAs included in TDS specification: · Description · Identification · Assay · Impurities and degradation products · Uniformity of dosage units · Permeation enhancer content, when
applicable · Adhesion · Release liner peel · Tack · Shear · Cold flow · In vitro drug release · Drug substance crystal presence · Pouch integrity · Microbial limits, when applicable · Moisture content, when applicable · Residual solvents |
The scope of the specification should comply with
Pharmacopoeial and relevant ICH guideline requirements, and should include
appropriate performance tests with respect to in vitro release / dissolution
and adhesion (see Section 4.2.6.1 In vitro drug release / dissolution and
Section 4.2.6.3.1 Adhesive Properties in vitro tests). The appearance of the
transdermal patch should also be fully specified. The limits should be in line with representative
batch and stability data, unless suitably qualified by non clinical, clinical
or other data. Limits for performance tests should be justified by
reference to clinical batches for which satisfactory efficacy and safety has
been demonstrated. The limits should be the same at release and shelf life,
unless justified and qualified by clinical data. |
||||||||||||||
|
申报批次生产要求 |
|
Applicants should submit data for registration/exhibit
batches manufactured from three distinct laminates, where each laminate is
made using different lots of drug substance, adhesives, backing, and/or other
critical elements in the TDS product. |
|
||||||||||||||
|
仿制药要求 |
研究者应当按照国家局发布的《化学仿制药参比制剂遴选与确定程序》选择参比制剂。仿制药的产品规格应当与参比制剂相同,关键质量特性也应不低于参比制剂。 仿制药与参比制剂相比,载药量和贴剂面积可能有所不同,但通常应具有相同或更高的贴剂面积活性(Patch Area Activity),且应确保在相同时间内递送的剂量相同,并应尽可能减少贴剂中的药物残留。 |
|
With respect to drug product quality, the following
elements should be compared: · Strength, as the mean dose delivered per
unit time, normally mass delivered in vivo per hour; · The content and location of active
substance in the drug product; · In vivo release rate or strength per patch
area (i.e. mass delivered in vivo/unit area/unit time); · Active substance utilisation (% of total
active substance absorbed per patient administration); · Patch area activity (active substance
utilisation/patch area); · Residual (mass of active substance
remaining in the drug product after completion of administration); · Instructions for use, including the use of
any overlay; and · Period of use. With respect to in vitro performance: Comparative drug release / dissolution, in vitro skin
permeation and adhesion / cohesive properties should be investigated and the
differences and similarities in in vitro performance between the generic and
reference products should be discussed, supported by appropriate data. For a generic or abridged application, the product
strength must be the same as the reference product. The other quality
elements, given above, should also be the same or similar, unless fully
justified. Clinical: To support a generic or abridged application,
bioequivalence with the reference product and also noninferiority (absence of
statistically significant difference) with respect to in vivo skin adhesion
should be demonstrated. Non-inferiority regarding clinical safety and local
tolerance of the generic product should also be demonstrated. |
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: