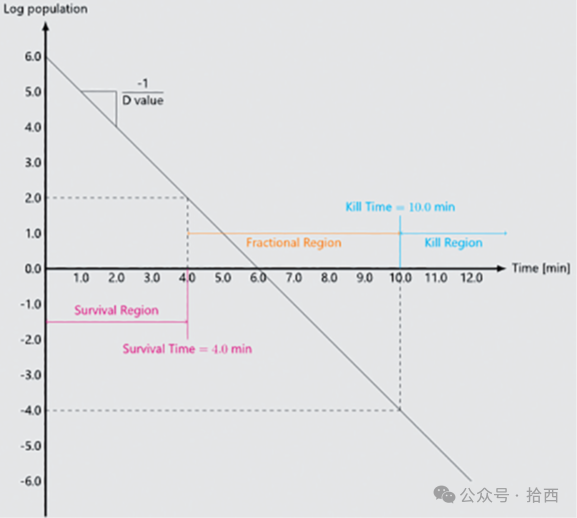
在制药行业无菌生产领域,过氧化氢生物净化工艺验证长期遵循着一个看似明确却暗藏隐患的金标准——生物指示剂完全杀灭。设备制造商提供的验证报告与药厂内部的周期性再确认,最终结论往往以"所有生物指示剂均呈阴性,实现≥6-log孢子杀灭"作为工艺合格的判定依据。然而,这一通行做法掩盖了一个根本性的科学缺陷:并非所有生物指示剂都具备相同的挑战特性。一个标称10⁶种群、D值0.7分钟的生物指示剂,与同样标称10⁶种群但D值2.0分钟的产品,所代表的生物学挑战强度存在本质差异。前者仅需约4.2分钟的等效作用即可达到6-log杀灭,后者则需要12分钟。然而在现行报告体系中,两者都会被简单记录为"6-log杀灭达成",仿佛代表了相同的工艺能力。这种信息不对称与透明度缺失,不仅可能导致不同系统、周期及供应商之间的误导性比较,更从根本上削弱了验证结果的科学严谨性。

生物指示剂质量变异性的实证基础
要理解为何仅报告"6-log杀灭"不够充分,首先需审视生物指示剂本身的质量状况。在近期Novak等发表的技术论文提供了极具说服力的基准数据,揭示了市售生物指示剂令人担忧的质量变异性。研究对多种产品进行了多年质量控制测试,发现产品间及批次间的变异性远超出微生物菌株间的差异。部分产品的质量控制实测种群值与分析证书标称值存在显著偏差;更为严重的是,在D值确认方面,实测值与证书值的差异有时竟达数百个百分点。许多批次因rogue发生率超过2%被判定为不合格——所谓rogue是指预期应完全杀灭的条件下仍呈阳性的异常抗性个体。这一数据揭示了一个令人不安的现实:未经严格入厂质量控制的使用者,实际上并不清楚自己正在使用何种特性的挑战工具。如果连验证工具本身的规格都不确定,验证结果的可靠性又从何谈起?这正是将种群数和D值纳入报告的根本出发点——只有当挑战工具的规格被明确记录并透明化,验证结果才具备可解释性和可比较性。
透明度框架——应当报告什么
基于上述分析,四个维度的信息被主张纳入到验证报告中。
种群数方面,虽然多数标称10⁶,但实际可能在5×10⁵到9×10⁶间波动,"完全杀灭"若对应实际种群仅5×10⁵,则只代表约5.7-log杀灭。报告实际种群使杀灭水平计算具有可追溯性。
D值方面,尽管不同供应商间因测试方法未标准化无法绝对比较,但对于同一供应商的同一产品线,批次间D值趋势可提供有价值的定性参考。当某批次D值显著偏离历史范围时,即便最终结果仍为"通过",也应重新评估工艺挑战强度。更重要的是,报告D值使验证结果具有"挑战强度"维度——使用低D值生物指示剂实现的6-log杀灭与使用高D值者,在生物学意义上截然不同。
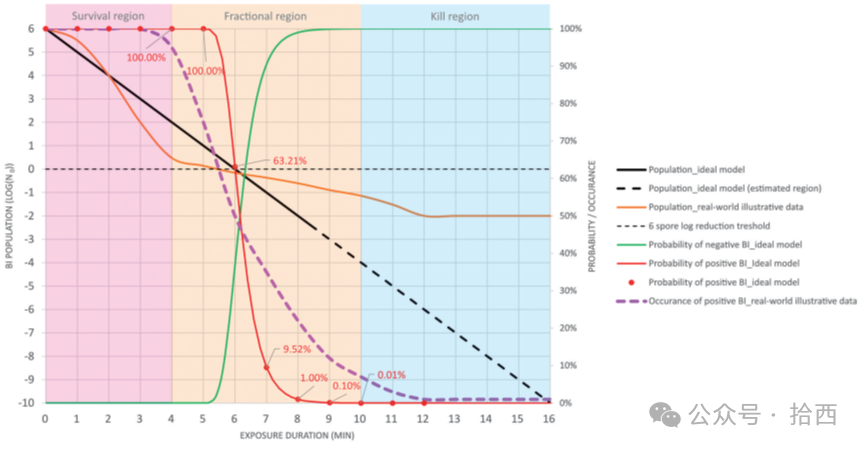
抗性特征方面,即灭活曲线是否接近理想模型、拖尾程度如何、rogue发生率多少,这一信息对于解释异常现象至关重要。若某次验证出现意外阳性而所用批次已知具有较高rogue发生率,则阳性更可能归因于生物指示剂而非工艺缺陷。
分离区杀灭数据方面,各暴露时间点的阳性率不仅可用于计算D值,还可评估批次的均一性与可预测性。理想情况下阳性率应随暴露时间平滑下降;异常波动或平台期则提示质量问题。
综合四维信息,报告可从简单表述升级为:"本次验证使用批次X,质量控制确认种群为1.2×10⁶,参考系统测定D值为1.8分钟,rogue发生率1.2%,灭活曲线呈轻微拖尾。在隔离器条件下所有BI均被灭活,等效杀灭≥6.1-log。"这样的报告不仅记录结果,更记录结果产生的完整背景。
孢子层的多层堆积可能导致底层孢子被物理屏蔽;孢子聚集体内部的个体可能因聚集体结构的保护而延迟暴露; 载体表面的微观不平整可能为个别孢子提供物理庇护所; 初级包装材料透气性的批次间波动可能造成局部暴露条件的不均一; 甚至不排除孢子群体中天然存在着极少数抗性显著高于群体平均水平的变异个体。
其一,通过扫描电子显微镜对大量生物指示剂进行成像,定量表征孢子层的厚度、均匀度与聚集程度,将这些形态学参数与rogue发生率进行系统的相关性分析。如果多层堆积确实是rogue的成因之一,则预期rogue发生率与局部孢子层厚度之间存在统计上显著的正相关关系。更进一步的验证可以通过控制实验来实现——人为制备单层分布与多层分布的标准样品,在相同暴露条件下比较两者的rogue发生率。 其二,考察载体材料与表面性质的影响。可将同批次孢子悬浮液分别接种至不同材质及不同表面粗糙度的载体上,比较各组的抗性分布与rogue发生率。如果载体表面的微观缺陷确实为孢子提供了物理庇护,则粗糙表面组的rogue发生率应显著高于光滑表面组。 其三,评估初级包装的贡献。可将同一批生物指示剂在不同封装条件下进行暴露比较,以检验包装材料透气性的批次间差异是否足以影响灭活曲线形态。 其四,从rogue个体中分离存活的单孢子,扩增培养后测定其抗性特征。如果rogue确实源于孢子群体中存在的天然高抗性变异体,则这些单孢子分离株应表现出显著高于亲本群体的D值。
标准化与可转移性的悖论
讨论生物指示剂抗性透明化时,必须正视标准化D值在汽化过氧化氢净化工艺中的实际意义。汽化过氧化氢工艺是一个气-液双相复杂过程,过氧化氢溶液蒸发后在腔内分布,在表面可能发生吸附和微冷凝,其灭活效果不仅取决于气相浓度,更与表面温度、相对湿度、材料吸附特性等众多因素密切相关。在此条件下,即使建立了标准化D值测试方法,所测D值也只能代表该BI在该特定测试系统中的抗性,无法直接外推至使用者自己的隔离器系统。隔离器的几何形状、装载模式、材料表面、环境条件均会影响过氧化氢分布与冷凝行为,从而改变BI的实际灭活动力学。
这一认识对验证实践具有深远的指导意义。首先,它意味着行业不应对跨供应商、跨产品的D值绝对可比性抱有过高的期望——不同供应商使用不同方法测得的D值本就不应直接比较。更为务实的做法是使用者建立自己的参考测试系统,对每批入厂的生物指示剂进行质量控制的相对评估,重点关注该批次相对于同一供应商历史批次的D值趋势与rogue发生率变化,而非追求与其他供应商产品之间的数值对标。其次,它意味着生物指示剂的最终适用性评判标准只能是其在具体隔离器工艺中的实际表现——任何实验室条件下获得的证书数据都无法取代在使用者自己的系统中进行的工艺挑战测试。然而,这绝不意味着D值报告没有价值。恰恰相反,正是因为汽化工艺的复杂性和不可外推性,在验证报告中透明化记录所使用的生物指示剂批次的参考系统D值、种群数和抗性特征才显得尤为重要——这些信息至少为验证结果提供了可追溯的生物学背景,使得不同时间、不同批次之间的比较具有了参照基准。当一个验证报告仅记录"6-log杀灭达成"而不提供任何关于挑战工具规格的信息时,该结果对后续研究者或检查员的参考价值极为有限;但当报告同时记录了所使用批次的实测D值与rogue水平时,该验证结果就具备了远为丰富的科学内涵。
综合以上论述,近期Novak等发表的技术论文中提出过氧化氢生物净化验证报告应从当前二元化的"通过/不通过"范式,转向基于数据透明化与风险量化的新范式。这一转变的核心并非单纯的技术进步,而是对验证本质的更深理解——验证的目的不应仅是证明某次循环达标,而是为整个生产周期内工艺的稳健性建立可追溯的科学基础。当验证仅记录"所有BI均被灭活"时,它提供的是静态快照;但当同时记录种群数、D值、抗性特征等多种数据时,它提供的是动态多维度的工艺画像。这一画像的价值在以下场景中尤为突出:同一隔离器再验证时,若两次使用BI批次抗性存在显著差异而结果均为"通过",我们需要知道第二次是否建立在更弱的挑战基础上;不同设计的两台隔离器比较时,若报告都写"6-log杀灭"但分别使用D值相差两倍的BI,其工艺能力真的可比吗?出现意外阳性时,若报告无rogue历史数据,调查将不得不在信息真空中进行。
值得注意的是,这种倡导的透明度框架与现有法规要求并不冲突。EU GMP Annex 1要求BI的入厂质量和抗性特征应被确认;USP<55>和ISO 11138-1也规定了种群计数与抗性测试要求。然而这些要求通常被视为供应商责任——供应商在分析证书上提供了数据,使用者似乎就完成了合规义务。然而,负责的使用者不应止步于此。分析证书数据描述的是"产品规格"而非"实际使用条件下的表现"。使用者应在验证报告中明确记录所使用批次的质量控制数据——无论来自供应商还是自己的入厂检测——以及这些数据如何支持对验证结果的解释。这种做法的额外成本主要涉及在报告中增加几个数据字段,但其科学严谨性与风险透明度提升却是显著的。更为重要的是,这种透明化报告建立了一种可追溯的验证文化——每一次验证不仅是为完成一次确认,更是积累关于BI批次间变异性和工艺稳健性的历史数据,使未来决策建立在更加充分的信息基础上。
结论:从"是否达标"到"如何达标"
作者:Shengyi
来源:拾西
公众号日期:2026年6月21日
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: